In quale Sindrome di Ehlers-Danlos, note anche come EDS, sono malattie del tessuto connettivo ereditate come parte di un difetto genetico. Soprattutto, l'EDS si manifesta attraverso le articolazioni ipermovibili e l'eccessiva elasticità della pelle. Occasionalmente, anche vasi, legamenti, muscoli, tendini e organi interni possono essere colpiti da EDS; la prognosi dipende dal tipo di sindrome diagnosticata.
Che cos'è la sindrome di Ehlers-Danlos?

© Astrid Gast - stock.adobe.com
La sindrome di Ehlers-Danlos è una malattia del tessuto connettivo molto rara. È una malattia ereditaria che si manifesta come parte di un difetto genetico. C'è un disturbo della sintesi del collagene. Poiché il tessuto connettivo è presente in tutto il corpo, i sintomi ei segni della sindrome di Ehlers-Danlos possono essere molto diversi e vari.
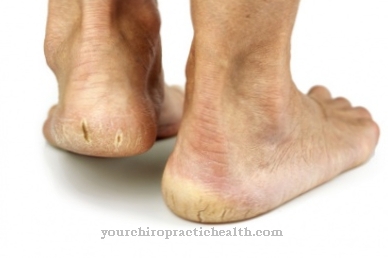
A volte la persona colpita può lamentarsi di una pelle troppo stirabile; in alcuni casi si parla addirittura di rottura dei vasi e degli organi interni. Esistono dieci tipi di EDS, che hanno sintomi e decorsi diversi.
cause
La sindrome di Ehlers-Danlos è scatenata da un difetto genetico nella sintesi del collagene. Circa l'80% di tutti i malati soffre di tipi da I a III, i tipi da IV a VII costituiscono solo una frazione di tutti i malati; I tipi da VIII a X sono ancora più rari. Mentre i tipi I e II della sindrome di Ehlers-Danlos influenzano il collagene V, il tipo III è un disturbo del collagene III.
I seguenti geni possono innescare la sindrome di Ehlers-Danlos: da COL1A1 a COL3A1 così come COL5A1 e COL5A2 e TNXB. A volte i geni ADAMTS2 e PLOD1 possono anche essere responsabili della sindrome. A seconda del tipo, dipende anche la modalità di ereditarietà. Molte forme di sindrome di Ehlers-Danlos sono ereditate come tratto autosomico dominante.
Ciò significa che solo uno degli alleli deve essere influenzato dal cambiamento affinché la sindrome di Ehlers-Danlos possa scoppiare. Ma ci sono anche forme che vengono ereditate come tratto autosomico recessivo. Entrambi gli alleli devono mostrare un cambiamento in modo che la malattia possa essere trasmessa.
Sintomi, disturbi e segni
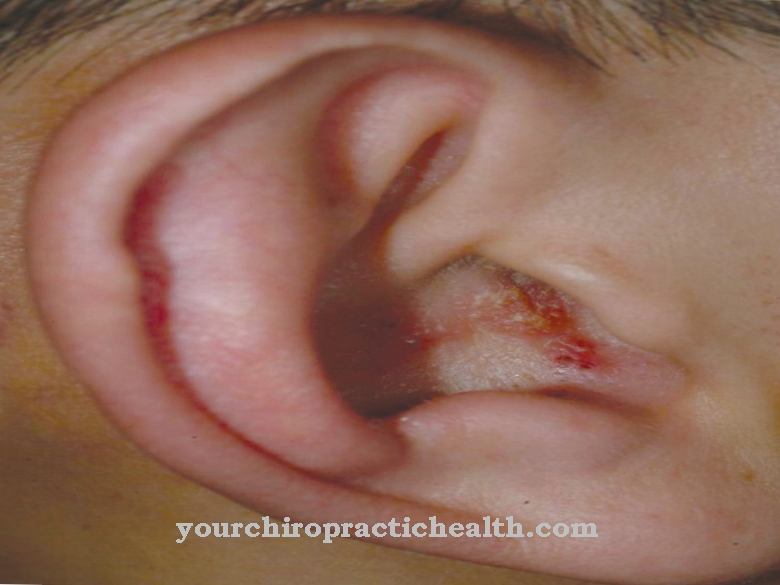
A causa del disturbo della sintesi del collagene, i vasi sanguigni, la pelle e le articolazioni non sono sufficientemente sviluppati. Ciò significa che il paziente soffre di una mancanza di fermezza e, di conseguenza, di un'eccessiva elasticità del tessuto connettivo. Questo porta ad un leggero strappo delle strutture già interessate. Soprattutto, i vasi sanguigni possono lacerarsi, si verificano curvature della colonna vertebrale o rotture intestinali. A volte è possibile anche uno pneumotorace ricorrente.
Con i tipi I e II, il paziente lamenta una pelle molto facilmente lesionabile e stirabile. Il processo di guarigione della ferita è molto anormale e le articolazioni sono molto forti e in alcuni casi possono essere colpiti anche i vasi sanguigni e gli organi interni.
Nel tipo III, la pelle è solo parzialmente interessata dal disturbo; Soprattutto, le persone colpite lamentano un'eccessiva mobilità delle articolazioni. Il tipo IV ha una pelle molto sottile, quasi traslucida. Il paziente ha una spiccata tendenza all'ematoma e ha anche articolazioni estremamente ipermovibili, per cui in questa fase i vasi e gli organi interni sono spesso anche colpiti dalla sindrome di Ehlers-Danlos.
Il tipo VI si lamenta principalmente di una moderata elasticità eccessiva della pelle. La guarigione della ferita è anormale; a volte sono coinvolti anche organi interni. In molti casi, c'è anche il coinvolgimento degli occhi. Nel tipo VII A e B, la pelle è estensibile, che a volte è estremamente sottile. Molti pazienti lamentano anche una lussazione dell'anca. Con il tipo VII C, invece, il paziente segnala rilassamento cutaneo e coinvolgimento dei suoi organi interni. Anche le articolazioni sono ipermovibili.
Diagnosi e corso
Il medico effettua una diagnosi clinica, facendo principalmente riferimento alla storia familiare. Il medico controlla anche l'aumento della fragilità capillare utilizzando il test di Rumpel-Leede ed esegue anche una biopsia cutanea, seguita da un esame al microscopio elettronico dell'intera struttura del collagene. I tipi possono essere identificati utilizzando l'amplificazione genetica del DNA umano.
A seconda del tipo, la prognosi e il decorso della malattia della sindrome di Ehlers-Danlos variano. Mentre alcuni pazienti hanno pochissime limitazioni, altri hanno problemi nella vita di tutti i giorni. Soprattutto, dolore, instabilità articolari estreme e deformazioni della colonna vertebrale significano che c'è un'enorme limitazione della mobilità.
La maggior parte dei pazienti, tuttavia, ha un'aspettativa di vita normale; Solo in pochissimi pazienti che soffrono anche di compromissione dei vasi sorgono complicazioni significative nel contesto della sindrome di Ehlers-Danlos.
complicazioni
Le complicanze nella sindrome di Ehlers-Danlos dipendono fortemente dal tipo diagnosticato. Nei casi più gravi, la pelle, i muscoli e i legamenti sono deformati. Anche gli organi interni possono essere colpiti. La pelle dei pazienti è spesso tesa eccessivamente e le articolazioni possono muoversi eccessivamente.
I bambini in particolare soffrono molto della sindrome di Ehlers-Danlos a causa del bullismo e delle prese in giro. Questo può portare a depressione e problemi psicologici.
La pelle può essere danneggiata anche da lievi influenze meccaniche ed è facile da strappare. Ciò aumenta il rischio di incidenti e infiammazioni.
La colonna vertebrale è anche interessata dalla sindrome di Ehlers-Danlos, che può portare a curvature della colonna vertebrale. Con tutti i tipi a volte c'è una cicatrizzazione anormale della ferita, quindi le infezioni e le infiammazioni devono essere trattate con particolare attenzione. Il trattamento o la terapia non sono possibili con la sindrome di Ehlers-Danlos.
Tuttavia, il paziente deve fare attenzione a non eseguire attività fisicamente faticose. Allo stesso modo, ferite e raffreddori comuni devono essere trattati con grande cura.La sindrome di Ehlers-Danlos non porta a una ridotta aspettativa di vita. Le complicazioni possono derivare principalmente dalla curvatura della colonna vertebrale o dalla guarigione ritardata delle ferite.
Quando dovresti andare dal dottore?
Un medico dovrebbe essere consultato per frequenti lacerazioni della pelle, cicatrizzazione anormale della ferita e altri sintomi indicativi di tessuto connettivo compromesso. La sindrome di Ehlers-Danlos si manifesta anche con ematomi, pelle eccessivamente sensibile e talvolta anche curvature della colonna vertebrale e rotture intestinali. Nel corso successivo possono verificarsi anche problemi all'anca e malattie degli organi interni. Per tutti questi reclami è necessario un chiarimento medico immediato.
Poiché la sindrome di Ehlers-Danlos è una malattia ereditaria, ha senso dare un'occhiata alla storia familiare. I genitori in attesa che soffrono personalmente della malattia o hanno casi della malattia nelle loro famiglie dovrebbero preferibilmente far esaminare il bambino immediatamente dopo la nascita.
Al più tardi quando si verificano gravi complicazioni come sanguinamento o forte dolore, un medico deve chiarire la causa. Altri contatti sono il dermatologo, vari internisti o uno specialista in malattie ereditarie. In caso di emergenza medica, è meglio allertare immediatamente il servizio di ambulanza o portare la persona colpita all'ospedale più vicino.
Medici e terapisti nella tua zona
Trattamento e terapia
Non esiste una terapia sintomatica o causale. Per questo tutto ruota intorno alla profilassi di eventuali danni consequenziali. Il paziente deve fare attenzione a evitare lesioni e sforzi fisici maggiori. Ciò significa che qualsiasi sport che a volte aumenta il rischio di lesioni dovrebbe essere evitato.
Il rischio aumenta durante la gravidanza; in particolare le persone affette dai tipi I, II, IV e VI devono quindi essere sottoposte a un attento monitoraggio. È anche importante avere una terapia antitosse per il raffreddore. Questo perché è possibile prevenire una rottura dell'intestino crasso e successivamente un pneumotorace. Se si verificano ferite, devono essere trattate con cura per incoraggiare la guarigione della ferita compromessa.
Outlook e previsioni
La prognosi per la sindrome di Ehlers-Danlos (EDS) dipende dal tipo di malattia. È anche diverso per ogni persona colpita. I pazienti con un tipo ipermobile di EDS di solito hanno un'aspettativa di vita normale. Con il tipo vascolare di EDS, c'è un maggior rischio di complicanze causate da un'emorragia interna improvvisa. In entrambe le forme, tuttavia, la qualità della vita è gravemente compromessa.
Inoltre, entrambe le forme di sindrome di Ehlers-Danlos contengono diverse malattie ereditarie del tessuto connettivo. Pertanto, al momento non è possibile alcuna terapia causale. Anche le opzioni per la terapia sintomatica sono limitate. È quindi importante che i pazienti facciano attenzione a non subire lesioni. Anche le operazioni banali non dovrebbero essere eseguite per evitare lesioni. Tuttavia, ci sono pazienti che difficilmente soffrono di limitazioni. Altri sono gravemente compromessi nella loro vita quotidiana.
Nel complesso, la sindrome di Ehlers-Danlos è progressiva. Nel corso della vita, le restrizioni aumentano. Possono verificarsi instabilità articolari, deformazioni spinali e forti dolori, che riducono notevolmente la mobilità. Nella forma vascolare della malattia, c'è anche un rischio costante che i vasi sanguigni si rompano, portando a una complicanza pericolosa per la vita. La peggiore conseguenza dell'EDS, tuttavia, è la grave compromissione della qualità della vita dovuta alla ridotta mobilità e alla costante paura di rotture dolorose.
prevenzione
Non c'è prevenzione. A volte solo gli esami come parte di una diagnosi preimpianto o una diagnosi del corpo polare possono prevenire l'ereditarietà della sindrome di Ehlers-Danlos.
Dopo cura
Con la sindrome di Ehlers-Danlos, la persona colpita di solito ha pochissime o addirittura nessuna misura di follow-up disponibile. Poiché si tratta di una malattia congenita, non può essere trattata in modo causale, ma solo sintomaticamente, in modo che non possa verificarsi una cura completa. Per questo motivo, la diagnosi precoce è in primo piano nella sindrome di Ehlers-Danlos in modo che non ci siano ulteriori complicazioni e reclami per la persona interessata.
Prima la sindrome viene riconosciuta da un medico, migliore è di solito l'ulteriore decorso. Se il paziente desidera avere figli, può essere eseguita anche la consulenza ereditaria per evitare che la sindrome venga trasmessa ai discendenti. Nella maggior parte dei casi l'aspettativa di vita del paziente non è influenzata negativamente da questa sindrome.
Tuttavia, le persone colpite dipendono molto dall'aiuto di amici e familiari nella loro vita quotidiana per essere in grado di far fronte alla vita quotidiana. Anche i vasi sanguigni dovrebbero essere controllati regolarmente in modo che non ci siano crepe, che possono essere pericolose per la vita della persona interessata. Nei bambini con sindrome di Ehlers-Danlos, dovrebbe essere fornita un'istruzione completa per evitare confusione.
Puoi farlo da solo
Il paziente non può curare la sindrome di Ehlers-Danlos con i suoi stessi aiuti. La malattia ereditaria è considerata incurabile. Nella vita di tutti i giorni, tuttavia, possono essere prese varie misure per alleviare i sintomi. Le opzioni di supporto devono essere utilizzate ripetutamente, poiché con esse non è possibile ottenere una minimizzazione permanente dei sintomi.
La debolezza del tessuto connettivo può essere nascosta da tagli alla moda in abiti e accessori. Indossare biancheria intima modellante e stabilizzante e allo stesso tempo indumenti un po 'larghi aiuta a evitare che le altre persone possano vedere i disturbi del tessuto connettivo nella vita di tutti i giorni. Massaggi, creme o bagni alternati stimolano la circolazione sanguigna nella pelle e sostengono il corpo. Le attività sportive aiutano a sviluppare i muscoli. In questo modo, alcune aree di debolezza del tessuto connettivo possono essere ridotte al minimo.
Genitori e medici dovrebbero spiegare in dettaglio la sindrome di Ehlers-Danlos e descriverne i sintomi e il decorso della malattia. Il bambino trae vantaggio dall'essere pienamente informato. Anche lo scambio digitale con altri malati può essere utile per affrontare la malattia nella vita di tutti i giorni. La fiducia nelle proprie competenze dovrebbe essere specificamente sostenuta e incoraggiata nelle prime fasi della vita del bambino. Avere una forte autostima aiuta il bambino ad avere una buona qualità di vita con la sindrome.
.jpg)
.jpg)





.jpg)



.jpg)