Del La malattia di Andersen è una forma particolarmente grave di malattia da accumulo di glicogeno, una condizione ereditaria caratterizzata dalla formazione di glicogeno anormale. La prognosi per la malattia è molto sfavorevole.
Cos'è la malattia di Andersen?

© ag visuell - stock.adobe.com
Nel contesto di La malattia di Andersen viene immagazzinata una forma insolita di glicogeno. Questo glicogeno ha una struttura simile all'amilopectina, una percentuale elevata della quale si trova nell'amido vegetale. Di solito il glicogeno è altamente ramificato. Nella malattia di Andersen, tuttavia, esiste solo un polisaccaride debolmente ramificato.
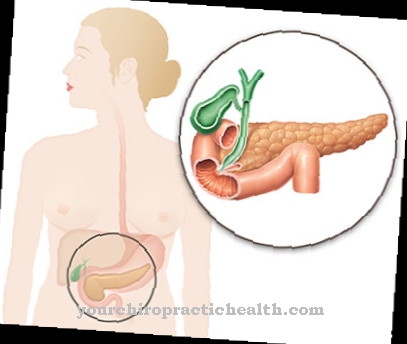
La malattia è caratterizzata da un rapido ingrossamento del fegato, che porta rapidamente alla cirrosi epatica. Il polisaccaride anormale non può più essere scomposto e continua ad accumularsi. La carenza o anche la mancanza dell'enzima amilo-1,4-1,6-transglucosidasi è responsabile della formazione difettosa del glicogeno. Fornisce la ramificazione in questa molecola di polisaccaride.
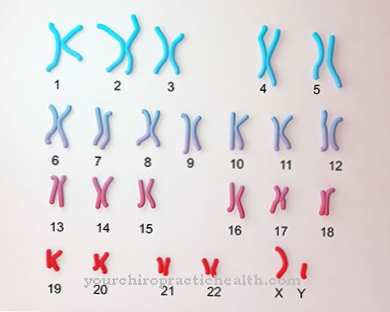
La malattia è molto rara, ma si manifesta ancora in varie forme o forme. Nella forma estremamente grave, il bambino è spesso nato morto. Sono state descritte anche forme più leggere che iniziano in età più avanzata. In ogni caso, tuttavia, è presente una mutazione nel gene (GBE1), che si trova sul cromosoma 3.
cause
La causa della malattia di Andersen è un difetto genetico nel gene GBE1 sul cromosoma 3, che può essere ereditato come tratto autosomico recessivo. Questo gene è responsabile della sintesi dell'enzima amilo-1,4-1,6-transglucosidasi. Se questo enzima manca o ha solo una funzionalità limitata, il normale glicogeno non può più essere sintetizzato. L'enzima è responsabile della ramificazione della molecola di polisaccarosio.
Se questa ramificazione non avviene o se viene eseguita solo in modo incompleto, si crea un glicogeno che non può più essere scomposto per un rapido apporto energetico. Al contrario, si accumula molto rapidamente nel fegato, nella milza e nei linfonodi. Dopo ogni pasto, parte del glucosio inutilizzato viene trasportato al fegato per immagazzinarlo come sostanza di riserva, il glicogeno.
Tuttavia, questo materiale di riserva non può essere utilizzato nella sua forma attuale. Il costante accumulo del glicogeno anormale allarga sempre di più il fegato e la milza e porta inevitabilmente alla distruzione di entrambi gli organi.
Sintomi, disturbi e segni
La malattia di Andersen si manifesta attraverso una straordinaria variabilità. Si tratta della conservazione costante di un glicogeno anormale, che non può più essere scomposto. Ma la gravità della malattia può essere diversa. Tuttavia, la prognosi per la malattia di Andersen è complessivamente molto sfavorevole. Il sintomo più evidente è un fegato in costante ingrandimento, da cui si sviluppa rapidamente la cirrosi epatica.
La forma più grave si manifesta attraverso i movimenti del bambino mancanti o ridotti prima della nascita. Il feto mostra segni di rigidità articolare e ipoplasia polmonare. Di solito in questi casi il bambino nasce morto. Nei casi classici, il bambino è ancora normalmente sviluppato alla nascita. Tuttavia, nei primi mesi di vita, si sviluppano epatomegalia (ingrossamento del fegato) e ipotonia (mancanza di tensione muscolare).
Nel complesso, lo sviluppo del bambino è ritardato. La malattia progredisce rapidamente. Il fegato sviluppa la cirrosi. C'è anche un aumento della pressione portale e la milza si ingrandisce. A causa della cirrosi epatica, le varici si sviluppano nell'esofago con corrispondente sanguinamento e ascite. La morte di solito si verifica nella prima infanzia. In casi più rari, la malattia inizia più tardi e mostra sintomi di debolezza muscolare e insufficienza cardiaca. Anche i sintomi neurologici si verificano qui.
Diagnosi e decorso della malattia
La diagnosi può essere effettuata sulla base del quadro clinico e accompagnata da esami di laboratorio, biopsie epatiche e test di genetica molecolare. Negli esami istologici è evidente l'accumulo intracellulare di strutture colorabili simili all'amilopectina. L'enzima responsabile viene esaminato negli epatociti, fibroblasti e leucociti. Una comprovata carenza di amilo-1,4-1,6-transglucosidasi conferma la diagnosi.
complicazioni
Di norma, l'aspettativa di vita del bambino è significativamente ridotta dalla malattia di Andersen o il bambino nasce morto. Questo può portare a gravi disturbi psicologici o depressione, soprattutto con parenti o genitori. Nella maggior parte dei casi, dipendono quindi dal trattamento psicologico.
I bambini affetti soffrono di cirrosi epatica, che alla fine porta alla morte. Inoltre, anche le articolazioni sono irrigidite e i movimenti non sono più possibili a causa di questa lamentela. Lo sviluppo mentale del bambino è anche gravemente compromesso dalla malattia di Andersen, quindi le persone colpite dipendono solitamente dall'aiuto di altre persone. Non è raro che si verifichino insufficienza cardiaca o debolezza muscolare.
I pazienti possono anche morire di morte cardiaca. Sfortunatamente, la malattia di Andersen non può essere curata. Il trapianto di un fegato può anche alleviare i sintomi solo per un breve periodo, poiché si verificherà anche il danno al nuovo fegato. Questo alla fine porta alla morte del bambino. Fino ad allora, tuttavia, i reclami ei sintomi possono essere limitati con l'aiuto di misure mediche.
Quando dovresti andare dal dottore?
La malattia di Andersen è una malattia genetica che, nei casi più gravi, può portare alla morte del feto nell'utero. Pertanto, le donne incinte dovrebbero cercare cure mediche non appena si notano irregolarità o anomalie durante la gravidanza. Se la futura mamma ha la vaga sensazione che qualcosa non va con il nascituro, dovrebbe consultare un medico. Se il neonato sopravvive nei primi giorni e settimane dopo la nascita, è necessario un medico non appena le peculiarità diventano evidenti nell'ulteriore corso dello sviluppo. In caso di debolezza muscolare o disturbi del movimento, consultare un medico.
I disturbi della crescita sono segni di una malattia esistente e devono essere chiariti. Le anomalie cardiache, le deformazioni del corpo e le discrepanze nel comportamento del bambino devono essere esaminate e trattate. In molti casi la malattia porta ad un ingrossamento degli organi. In questi casi, il fegato o la milza sono particolarmente colpiti.
Pertanto, un medico è necessario non appena si verifica una forma insolita della parte superiore del corpo rispetto a neonati o bambini della stessa età. Lo scolorimento della pelle o altre irregolarità nell'aspetto della pelle sono ulteriori segni di danni alla salute. Un viso o occhi giallastri dovrebbero essere valutati da un medico.
Terapia e trattamento
Poiché la malattia è genetica, non può essere somministrato alcun trattamento causale. La terapia è solo sintomatica. Come parte del trattamento, i medici si concentrano principalmente sulle complicazioni che si presentano. Questo abbassa la pressione nel circuito della vena porta. C'è anche una sostituzione di albumina e fattori di coagulazione.
Se si verifica un'insufficienza epatica, un trapianto di fegato può prolungare la vita. Tuttavia, la malattia non può essere curata nemmeno con un trapianto di fegato. Il difetto genetico è presente e porterà anche a depositi di glicogeno anormale nel nuovo fegato. La conservazione del polisaccaride difettoso continua negli altri organi del cosiddetto sistema reticoloistiocitario della milza e dei linfonodi, cosicché possono ancora verificarsi gravi complicazioni anche dopo un trapianto di fegato riuscito.
Il sistema reticoloistiocitico fa parte del sistema immunitario e comprende le cellule del tessuto connettivo reticolare. Queste cellule immagazzinano particelle e sostanze per scomporle e poi portarle fuori dal corpo. Tuttavia, anche qui non è più possibile la scomposizione delle molecole di polisaccarosio difettose.
Outlook e previsioni
La malattia di Andersen ha una prognosi relativamente sfavorevole. La malattia metabolica non è stata finora curabile e causa gravi danni al fegato. In alcuni casi si verificano disturbi muscolari e malattie concomitanti che, se non trattate, progrediscono progressivamente. L'aspettativa di vita è fortemente limitata dalla condizione. I bambini malati raggiungono in media dai due ai cinque anni di età. Un trapianto di fegato precoce migliora la prognosi. La prognosi è sfavorevole, soprattutto per le forme classiche di malattia, soprattutto se nei primi mesi di vita non è previsto il trapianto di fegato.
Di regola, la prognosi a lungo termine si basa sull'estensione, la gravità e la progressione della malattia. La malattia di Andersen è una delle glicogenosi più gravi. La qualità della vita è generalmente notevolmente ridotta a causa di problemi al fegato e altri sintomi. I farmaci antidolorifici e la terapia completa migliorano il benessere del bambino, ma sono anche associati a rischi. Lo specialista del fegato responsabile fornisce la prognosi.
L'aspettativa di vita è fortemente limitata dalla condizione. Anche eventuali malattie concomitanti che possono verificarsi con malattie non rilevate sono incluse nella prognosi. La malattia di Andersen offre quindi una prognosi complessivamente sfavorevole. Nuovi metodi di trattamento potrebbero portare un miglioramento in futuro.
prevenzione
Una prevenzione della malattia di Andersen può solo riferirsi al fatto che la prole non eredita questa malattia. Poiché la malattia di Andersen viene trasmessa in modo autosomico recessivo, diverse generazioni possono essere ignorate in eredità. Se ci sono già stati casi di malattia di Andersen in famiglia e parenti, è necessario quindi eseguire test genetici umani.
Se il gene si trova in entrambi i genitori, si consiglia la consulenza genetica. In questo caso, la prole ha una probabilità del 25% di sviluppare la malattia di Andersen.
Dopo cura
Poiché la malattia di Andersen non può essere curata, il trattamento dei sintomi e il contenimento di possibili complicanze sono l'obiettivo principale per tutto il periodo di trattamento. La cura di follow-up è necessaria dopo gli interventi effettuati come parte della terapia. Se si verifica un trapianto di fegato, è molto importante un follow-up professionale.
Dopo la procedura, questo garantisce che il nuovo fegato non venga rifiutato dall'organismo. Farmaci speciali sopprimono la risposta immunitaria del corpo. Di conseguenza, tuttavia, la resistenza del corpo agli agenti patogeni viene indebolita, cosa che deve essere presa in considerazione in ulteriori terapie. Durante questo periodo, il paziente deve sottoporsi a regolari esami del sangue. Viene prestata attenzione per garantire che non vi siano reazioni di rigetto o altre gravi complicazioni come la disfunzione renale, che possono manifestarsi come effetti collaterali.
Mentre i sintomi principali della malattia di Andersen possono essere migliorati direttamente dopo un trapianto di fegato, la deposizione del glicogeno difettoso continua, quindi è necessario aspettarsi complicazioni e sintomi progressivi anche dopo un trapianto. Lo specialista epatico responsabile può fornire informazioni più dettagliate sulla prognosi e sull'ulteriore corso del trattamento.
Puoi farlo da solo
Le misure di auto-aiuto che un paziente con malattia di Andersen può prendere sono limitate a inesistenti. Poiché la malattia ha cause genetiche e non può essere controllata nonostante il trattamento sintomatico, le possibilità della persona colpita si esauriscono rapidamente. Si consiglia di prendere sul serio qualsiasi consiglio sulla dieta e sullo stile di vita fornito dal suo medico curante e di metterlo in pratica.
Inoltre, dopo un trapianto di fegato, la persona colpita dovrebbe considerare un comportamento gentile. Alcol, cibi grassi e attività fisica dovrebbero essere evitati. Questo rende più facile per il corpo accettare veramente il nuovo organo. Tuttavia, un trapianto di successo, comprese le cure di follow-up di successo, non può fermare la glicogenosi di tipo 4 stessa.
Poiché si tratta di una malattia ereditaria autosomica recessiva (può saltare diverse generazioni), ha senso avere un profilo genetico redatto quando si pianifica una famiglia. Mentre le persone colpite dalla malattia di Andersen conoscono già il loro gene, un'analisi a questo proposito è particolarmente utile per i membri della famiglia. In questo modo, la trasmissione del gene scatenante può essere prevenuta attraverso un'adeguata pianificazione familiare. Almeno, tuttavia, si può ottenere una certezza sul rischio di malattia nella propria prole.

.jpg)
.jpg)



.jpg)




.jpg)