Il Terapia enzimatica sostitutiva è utilizzato per trattare le malattie da accumulo lisosomiale, in cui la mancanza di enzimi porta ad un accumulo patologico di prodotti di degradazione nei lisosomi delle cellule.
Gli enzimi mancanti a causa di difetti genetici sono compensati da infusioni endovenose regolari. Poiché gli enzimi sintetici infusi non possono attraversare la barriera emato-encefalica a causa delle loro dimensioni molecolari, la terapia funziona solo per le malattie da accumulo lisosomiale che non colpiscono il sistema nervoso centrale.
Cos'è la terapia enzimatica sostitutiva?

I lisosomi sono organelli cellulari speciali in cui le sostanze estranee ed endogene vengono scomposte e parzialmente riciclate. Specifici enzimi idrolizzanti sono necessari per la degradazione e il trasporto delle sostanze. Queste sono proteasi, nucleasi, lipasi e sostanze trasportatrici.
Una serie di difetti genetici noti possono portare al fallimento di alcuni enzimi, per cui alcuni prodotti di degradazione si accumulano nei lisosomi in quantità patologiche e si accumulano fino a raggiungere la matrice extracellulare, cioè gli spazi intercellulari, in maniera incontrollata. Tutti i difetti genetici che portano al fallimento di almeno una idrolasi necessaria sono riassunti sotto il termine malattia da accumulo lisosomiale. La terapia enzimatica sostitutiva (ERT, terapia enzimatica sostitutiva) viene utilizzato per sostituire gli enzimi endogeni mancanti con enzimi prodotti sinteticamente.
Poiché le idrolasi sono costituite da molecole relativamente grandi, non possono essere assorbite dall'intestino senza prima essere scomposte e inattivate, in modo che possano essere somministrate solo tramite infusione endovenosa. Tuttavia, la dimensione delle molecole enzimatiche impedisce anche l'attraversamento della barriera ematoencefalica, in modo che la terapia possa essere efficace solo per le malattie da accumulo lisosomiale che non colpiscono il sistema nervoso centrale (SNC).
Funzione, effetto e obiettivi
Sono noti oltre 50 differenti disordini metabolici lisosomiali, ognuno dei quali riconducibile a un difetto monogenetico. Le malattie da accumulo lisosomiale possono essere suddivise in sette classi differenti a seconda delle sostanze eccessivamente immagazzinate a causa del difetto enzimatico esistente.
Le mucopolisaccaridosi e le oligosaccaridosi sono principalmente adatte per una ERT. Lo scopo di ERT è sempre quello di compensare la carenza enzimatica specifica attraverso gli enzimi forniti artificialmente al fine di portare la malattia a un punto morto o almeno a un decorso più lieve. In dettaglio, sono disponibili enzimi sostitutivi per le seguenti malattie da accumulo lisosomiale:
- Malattia di Gaucher
- Malattia di Pompe
- Malattia di Fabry
- Sindrome di Hurler-Pfaundler (mucopolisaccaridosi I)
- Malattia di Hunter (mucopolisaccaridosi II)
• Sindrome di Maroteaux-Lamy (mucopolisaccaridosi VI) • Niemann-Pick B
La malattia di Gaucher è la più comune malattia da accumulo lisosomiale. Si presenta in tre diverse varianti, due delle quali interessano anche il sistema nervoso. Nella forma non neuropatica, la milza è particolarmente colpita, che si ingrandisce notevolmente e porta a danni secondari come anemia e danni al midollo osseo. I sintomi tipici sono dolore alle ossa e alle articolazioni e disturbi circolatori. La variante neuropatica acuta della malattia mostra un decorso grave e offre poche possibilità di sopravvivenza oltre i primi due anni di vita.
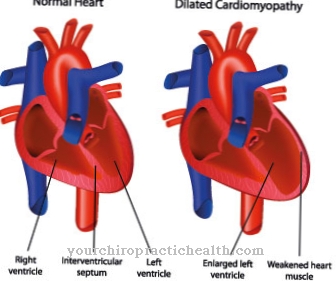
La malattia da accumulo La malattia di Pompe è dovuta a una carenza dell'enzima alfa-1,4-glucosidasi, che è coinvolto in un gran numero di processi metabolici. La malattia di Pompe porta a un enorme ingrossamento del cuore (cardiomegalia) e insufficienza cardiaca. Ci sono corsi precoci, gravi, che compaiono nei primi mesi di vita, così come forme più lievi che compaiono solo negli anni successivi di vita.
La malattia di Fabry è causata da un difetto genetico legato all'X, quindi solo i ragazzi e gli uomini possono essere colpiti dalla malattia da accumulo. La malattia di solito porta a sintomi nell'infanzia avanzata, inclusi attacchi di dolore, cheratomi della pelle, problemi renali e danni ai muscoli cardiaci. La carenza dell'enzima alfa-galattosidasi A porta ad un accumulo di ceramide triesoside, che è la causa dello scatenamento dei sintomi, che possono interessare anche il sistema nervoso autonomo.
Non è raro che il danno provochi un attacco di cuore, un infarto renale o persino un ictus. La sindrome di Hurler-Pfaundler è anche nota come mucopolisaccaridosi, tipo I ed è causata da un'interruzione del metabolismo dei glicosaminoglicani. La malattia è associata a un'ampia varietà di sintomi, inclusi gravi disturbi mentali e gravi alterazioni scheletriche. Il decorso della malattia è grave, quindi l'aspettativa di vita media è compresa tra 11 e 14 anni. La malattia di Hunter corrisponde alla mucopolisaccaridosi, tipo 2 ed è, come la malattia di Hurler, causata da un difetto legato all'X. La malattia è caratterizzata da decorsi di varia gravità, da quelli che si verificano nella prima infanzia a quelli lievi che compaiono solo negli uomini adulti.
A causa dei sintomi cardiaci più comuni come difetti delle valvole cardiache e problemi al muscolo cardiaco, l'aspettativa di vita varia da normale a leggermente limitata. La sindrome di Maroteaux-Lamy (MPS VI) è una delle mucopolisaccaridosi ereditate come tratto autosomico recessivo perché il difetto genetico che la causa non è sul cromosoma X. La malattia è molto rara, con un caso ogni 455.000 nati. Sono note forme lievi e gravi.
I sintomi sono fegato e milza ingrossati, sindrome del tunnel carpale e alterazioni delle valvole cardiache. Il Niemann-Pick B è una lipidosi della sfingomielina, che è una delle malattie da accumulo lisosomiale ed è causata da un difetto genetico sul cromosoma 11. Mentre il tipo B della malattia colpisce principalmente il fegato e la milza, il tipo A ha anche notevoli problemi neuronali.
Puoi trovare i tuoi farmaci qui
➔ Medicinali per il doloreRischi, effetti collaterali e pericoli
Poiché molte delle malattie da accumulo lisosomiale che possono essere trattate con la terapia enzimatica sostitutiva seguono un decorso grave con un tasso di mortalità corrispondentemente più elevato se non trattate, il rischio maggiore in ERT è che l'enzima sostitutivo selezionato non funzioni o funzioni solo troppo debolmente.
Un altro rischio risiede meno nella terapia stessa che nel fatto che la malattia sottostante viene riconosciuta troppo tardi, in modo che l'ERT possa interrompersi nel corso del decorso, ma il danno già causato non può regredire. Circa un paziente su due trattato reagisce temporaneamente alle infusioni con sintomi quali febbre e brividi. Le ragioni di ciò non sono ancora del tutto comprese. Alcuni pazienti reagiscono formando anticorpi e ci sono stati casi noti in cui i pazienti hanno reagito con eruzioni cutanee e broncospasmo.






.jpg)




.jpg)