Ce ne sono 45 diversi in totale malattie da accumulo lisosomiale, che sono un gruppo eterogeneo di malattie metaboliche congenite. Le persone che soffrono di una di queste malattie hanno un difetto genetico. Tutte le malattie da accumulo hanno una cosa in comune: un certo enzima è assente o solo parzialmente funzionante.
Cos'è la malattia da accumulo lisosomiale?

© designua - stock.adobe.com
Queste malattie da accumulo congenito sono rare, poiché ne sono colpite meno di cinque persone su 10.000. Le varie malattie hanno un decorso molto diverso ei sintomi possono variare ampiamente.
Le forme più famose di malattia da accumulo lisosomiale sono la malattia di Fabry, la malattia di Gaucher, la malattia di Pompe e la mucopolisaccaridosi (MPS). Spesso vengono definiti "orfani della medicina" perché la strada per una diagnosi specifica e una terapia adeguata può essere molto lunga. A volte possono volerci anni prima che le persone colpite scoprano cosa sta succedendo loro.
cause
Le malattie da accumulo lisosomiale sono caratterizzate da alcune forme di malattie metaboliche ereditarie. Ai pazienti manca un enzima importante che assicura che l'equilibrio metabolico funzioni senza intoppi. Nella forma meno pronunciata, questo enzima non è almeno presente in quantità sufficienti.
Il compito degli enzimi è quello di smaltire gli inquinanti e i materiali di scarto che si accumulano nell'organismo umano attraverso il metabolismo attraverso i lisosomi, o di elaborarli nuovamente in modo tale che i sintomi non si manifestino.
In caso di carenza enzimatica, questo ciclo di smaltimento senza problemi non è più garantito. Le sostanze nocive si insediano nelle cellule e interrompono il ciclo metabolico. Nella fase iniziale, i disturbi non hanno un effetto evidente, ci sono solo alcune restrizioni. Tuttavia, se questo disturbo metabolico rimane non trattato a causa di una carenza enzimatica, i sintomi si moltiplicano perché le cellule si ingrandiscono molto.
Sintomi, disturbi e segni
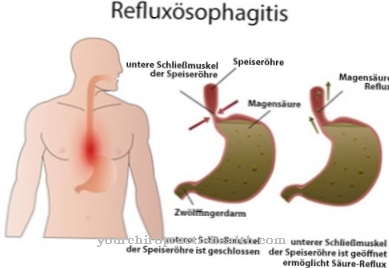
Nel peggiore dei casi, questi andranno sotto. Le conseguenze sono danni alle ossa, al sistema nervoso, alla milza, ai reni, ai muscoli o al cuore. La malattia di Fabry causa l'accumulo di grasso (globotriaosilceramide, Gb3) nelle cellule a causa della ridotta o assente attività enzimatica. Questi depositi indesiderati possono causare forti dolori alle dita dei piedi o delle dita, ictus e danni ai reni.
Diagnosi e decorso della malattia
Questo quadro clinico interessa diversi sistemi contemporaneamente: vasi sanguigni, reni, cuore e sistema nervoso. La malattia di Gaucher ereditaria autosomica recessiva causa una mutazione dell'enzima "beta-glucocerebrosidasi" e porta ad un accumulo di substrato all'interno delle cellule, soprattutto nei macrofagi (cellule scavenger), che appartengono al sistema reticolo-endoteliale. L'emocromo cambia, il fegato e la milza sono ingrossati e le ossa fanno male.
La malattia è progressiva ed è per lo più etnica, poiché nella maggior parte dei casi si manifesta in persone di origine ebraica. La malattia di Pompe è anche nota come "deficit di maltasi acida". Il quadro clinico appartiene al gruppo della glicogenesi di tipo II Le persone affette mancano dell'enzima "alfa-1,4-glucosidasi" (maltasi acida) o non è disponibile in quantità sufficienti. A causa della ridotta degradazione del glicogeno nei muscoli, i pazienti soffrono della distruzione delle cellule muscolari sotto forma di accumulo di zucchero.
La mucopolisaccaridosi di tipo I (MPS), nota anche come malattia di Hunter, ha varie cause cliniche. La malattia di Hurler è la forma più grave e la malattia di Scheie è alla fine della patogenesi clinica. Esistono transizioni di caratteristiche diverse tra queste due forme di progressione. La caratteristica più evidente è la ridotta scomposizione dei carboidrati che si accumulano nei lisosomi delle cellule.
I pazienti con malattia di Hunter possono manifestare bassa statura, ingrossamento della milza e del fegato, tratti ruvidi, pelle ispessita, lingua ingrossata e difficoltà respiratorie. Inoltre, lo scheletro viene spesso modificato nell'area del bacino, della colonna vertebrale, delle ossa delle mani e del cranio. Sono possibili ombelicali e [[ernie inguinali].
complicazioni
Nella maggior parte dei casi, i sintomi o le complicazioni compaiono molto tardi in questa malattia. Per questo motivo, viene diagnosticato in ritardo, quindi nella maggior parte dei casi non è possibile un trattamento precoce. Senza trattamento, con il progredire della malattia si verificano vari disturbi e danni agli organi interni.
I reni, il fegato e la milza sono particolarmente colpiti. Anche il cuore può essere colpito da questa malattia, che nel peggiore dei casi può portare alla morte cardiaca. Inoltre, si verificano danni ai reni e le persone colpite spesso soffrono di dolore alle dita dei piedi o delle dita. La paralisi può verificarsi anche se il cervello è stato danneggiato da questa malattia. Il fegato e la milza possono essere ingrossati e causare anche forti dolori.
Non è raro che le ossa della persona colpita siano fragili e anche dolorose. Il trattamento di questa malattia si rivela difficile. In molti casi, l'aspettativa di vita della persona colpita è notevolmente ridotta. Di solito non ci sono complicazioni particolari quando si usano i farmaci. Tuttavia, un decorso positivo della malattia non può essere garantito in ogni caso.
Puoi trovare i tuoi farmaci qui
➔ Medicinali per il doloreQuando dovresti andare dal dottore?
Perdita di capelli, problemi articolari e disturbi d'organo sono possibili segni di malattia da accumulo lisosomiale. Si consiglia una visita da un medico se i sintomi continuano a ripresentarsi o se compaiono all'improvviso senza che sia stata trovata una causa. Se i sintomi sono correlati a un difetto enzimatico già diagnosticato o ad altre malattie gravi, consultare il medico responsabile. Una malattia da accumulo non trattata può portare a demenza, infertilità, neuropatie e altre complicazioni, alcune delle quali pericolose per la vita. Pertanto, tutti i sintomi concepibili dovrebbero essere esaminati, anche se non vi è alcun sospetto concreto.
I sintomi della malattia da accumulo lisosomiale possono manifestarsi in fasi o svilupparsi in modo insidioso, ma richiedono sempre esame e trattamento. Le persone colpite sono le migliori per parlare direttamente con il loro medico di famiglia o un internista. La terapia vera e propria di solito avviene in una clinica specialistica per malattie interne, anche se la fisioterapia o la psicoterapia possono essere collegate a seconda dei sintomi. In particolare, sono indicate misure terapeutiche a causa del decorso spesso negativo della malattia.
Terapia e trattamento
A seconda di quanto precocemente viene fatta una diagnosi adeguata, queste malattie ereditarie possono essere trattate molto bene con la terapia enzimatica sostitutiva, in modo che le persone colpite abbiano molti meno sintomi e quindi una migliore qualità della vita. Questa terapia sostitutiva viene utilizzata in base al quadro clinico.
Le persone che soffrono della malattia di Gaucher mancano dell '"enzima ß-glucocerebrosidasi", che viene prodotto biotecnologicamente e infuso nel corpo del paziente. I lisosomi agiscono in modo efficiente e sono in grado di assorbire sostanze dal loro ambiente immediato. Per questo motivo, gli enzimi utilizzati artificialmente vengono modificati in modo tale da poter essere forniti ai lisosomi in modo ideale.
I macrofagi (fagociti) scompongono i glucocerebrosidi che si sono accumulati nelle cellule. Questa terapia può essere paragonata alla terapia insulinica per il diabete mellito, con la differenza che non è un ormone mancante, ma un enzima non disponibile. Il corpo scompone regolarmente tutte le sostanze, compreso l'enzima artificiale fornito.
A causa di questa regolare scomposizione della sostanza, i pazienti devono sottoporsi regolarmente a questa terapia infusionale fino alla fine della loro vita. La terapia enzimatica sostitutiva non agisce in modo sintomatico, ma combatte direttamente la causa della malattia ereditaria. I medici chiamano questa terapia causale. I principi della terapia devono essere utilizzati per tutte e quattro le malattie da accumulo comuni summenzionate.
Anche i pazienti Pompe sono trattati con terapia infusionale. In questa malattia, viene fornito l'enzima inesistente "acido alfa glucosidasi" che aiuta ad abbattere il glicogeno che si è accumulato nei lisosomi dei muscoli. Nei pazienti con malattia di tipo “mucopolisaccaridosi di tipo I” l'enzima lisosomiale “alfa-iduronidasi” non è presente o non è presente in quantità sufficienti. È una delle malattie da accumulo più rare in cui le molecole di zucchero si accumulano negli organi e nei tessuti.
Se il processo è normale, l'enzima scompone i mucopolisaccaridi. Le molecole di zucchero sono a catena lunga e sono coinvolte nello sviluppo del tessuto di supporto e connettivo, ad esempio ossa, pelle, fluidi articolari e cartilagine. Se il normale decorso della degradazione è disturbato a causa della mancanza di enzimi, i glicosaminoglicani patologici (GAG) si accumulano nelle singole cellule. Le future opzioni terapeutiche mirano all'assunzione di compresse.
Outlook e previsioni
La prognosi per la malattia da accumulo è infausta. Si è scoperto che una predisposizione genetica è la causa del disturbo di salute. I requisiti legali vietano a medici e scienziati di modificare la genetica umana. Per questo motivo, la malattia rimane per tutta la vita e non ha prospettive di guarigione.
Il medico curante si concentra sul trattamento dei sintomi che si presentano. Se non vengono trattati, vari reclami aumenteranno nel tempo. Il sistema osseo è danneggiato e sorgono problemi agli organi. Nel peggiore dei casi, ci saranno difficoltà funzionali negli organi interni e alla fine un fallimento dell'attività dell'organo. Questo minaccia la persona interessata di morte prematura.
La sfida della malattia sta nella diagnosi. In un gran numero di pazienti, i sintomi significativi e fortemente percepibili compaiono solo più tardi nella vita. Di conseguenza, la malattia genetica rimane inosservata per molto tempo e il trattamento precoce della malattia è difficile. Più tardi viene fatta una diagnosi, più sfavorevole è l'ulteriore corso. In una fase avanzata della malattia, gli organi interni o le articolazioni sono già gravemente danneggiati. Sono necessari interventi chirurgici e se la malattia progredisce male, solo un organo donatore può salvare la vita della persona colpita. Il trattamento precoce è quindi essenziale per una prognosi migliore.
prevenzione
Trattandosi di un difetto genetico congenito che impedisce l'espressione di un enzima, questa malattia non può essere trattata preventivamente. Tuttavia, gli ultimi risultati dell'ingegneria genetica potrebbero fornire un approccio in questo campo.
Dopo cura
Con questa malattia, le persone soffrono di una serie di diverse complicazioni e disturbi. Di norma, tutti questi hanno un effetto molto negativo sulla qualità della vita della persona colpita, quindi una diagnosi dovrebbe essere fatta molto presto. Prima viene consultato un medico, migliore è di solito l'ulteriore decorso di questa malattia.
La gravità di questa malattia può essere molto diversa, quindi una previsione generale spesso non è possibile. Le persone colpite soffrono di gravi danni agli organi interni. I reni e il cuore sono principalmente colpiti, in modo che il bambino possa morire nei primi giorni se i sintomi non vengono corretti in tempo. Ci sono anche depositi di grasso in diverse parti del corpo.
Le dita delle mani e dei piedi sono particolarmente colpite, il che può portare a un'estetica notevolmente ridotta per la persona colpita. Di regola, i reni e il cervello vengono danneggiati nel corso successivo, quindi la persona interessata muore a causa di questo danno. Anche i genitori e i parenti soffrono spesso di depressione o altri disturbi mentali dovuti alla malattia.
Puoi farlo da solo
Le malattie da accumulo lisosomiale molto spesso richiedono cure mediche intensive. Spesso non ci sono abbastanza opportunità per l'auto-aiuto. I genitori dei bambini colpiti spesso sperimentano un forte stress nel loro ambiente domestico perché il loro bambino ha bisogno di cure e attenzioni costanti.
I quadri clinici delle singole malattie da accumulo sono differenti. Esistono sia forme facili che molto difficili. Un esempio è la malattia di Gaucher. L'aiuto dei genitori è spesso limitato all'alimentazione del bambino gravemente disabile. Nei casi più lievi, l'aspettativa di vita può essere quasi normale. Tuttavia, è necessaria una costante supervisione medica per evitare possibili complicazioni. L'attività fisica regolare è una delle terapie di accompagnamento che possono essere svolte anche a casa. Inoltre, deve essere organizzato un attento esame di screening del cancro. Ciò richiede visite costanti dal medico con il loro bambino dai genitori. Lo stesso vale per altre malattie da accumulo lisosomiale.
In caso di alcune malattie, oltre alle disabilità fisiche, possono verificarsi anche limitazioni mentali, che richiedono comunque un sostegno speciale. Nelle forme più lievi di alcune malattie, come la malattia di Hunter, si verificano inizialmente solo alterazioni scheletriche e dismorfismi facciali. Qui, tuttavia, il paziente colpito è spesso in grado di condurre una vita indipendente. Tuttavia, anche qui sono necessari esami medici costanti per escludere possibili complicazioni come insufficienza cardiaca o malattie respiratorie. Il paziente può affrontare lo stress psicologico causato da deformazioni fisiche attraverso la consulenza psicologica.
.jpg)