Il Sindrome di Allan-Herndon-Dudley è una mutazione nel gene SLC16A2 che cambia il trasportatore dell'ormone tiroideo MCT8 e causa una ridotta captazione di iodotironina nel tessuto muscolare e nel sistema nervoso centrale. A causa della mutazione, le persone colpite soffrono di debolezza muscolare e ritardi nello sviluppo mobile e mentale. L'AHDS è incurabile e finora è stato trattato solo con la somministrazione di triiodotiroacetato.
Che cos'è la sindrome di Allan-Herndon-Dudley?

I ritardi nello sviluppo fisico, mentale o emotivo di adolescenti e bambini sono riassunti come ritardi o ritardi dello sviluppo. I ritardi nello sviluppo possono avere cause diverse. Ad esempio, il fattore scatenante per lo sviluppo ritardato può trovarsi nel sistema nervoso centrale.
È il caso, ad esempio, della sindrome di Allan-Herndon-Dudley (AHDS). Oltre al grave ritardo mentale, la sindrome è caratterizzata da disturbi dello sviluppo motorio. La prima descrizione della sindrome risale al 1944. Il quadro clinico che descrivi è una malattia ereditaria, cioè una malattia genetica.
La malattia colpisce i neonati maschi nella maggior parte dei casi documentati fino ad oggi. I disturbi dello sviluppo e le loro conseguenze sono in quasi tutti i casi chiaramente visibili dalla nascita. L'AHDS è una malattia estremamente rara. Per questo motivo, lo stato della ricerca sulla sindrome di Allan - Herndon - Dudley è stato finora piuttosto scarso.
cause
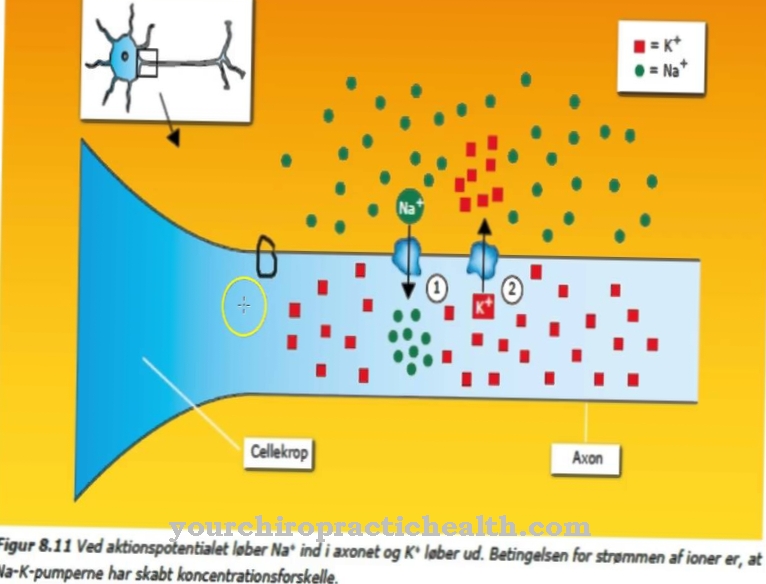
L'AHDS è una malattia genetica ereditaria causata da una mutazione nel gene SLC16A2. Questo è il gene codificante per il cosiddetto trasportatore dell'ormone tiroideo MCT8. Questo trasportatore media l'assorbimento di iodotironine nel tessuto muscolare e nervoso.
A causa della mutazione, si verificano disturbi durante l'assorbimento degli ormoni tiroidei, che squilibrano il sistema nervoso centrale e quindi compromettono lo sviluppo delle cellule del sistema nervoso. Il tessuto muscolare e il cervello si impoveriscono a causa della disregolazione correlata alla mutazione dell'ormone tiroideo attivo da cui dipendono effettivamente.
La sindrome viene trasmessa come eredità recessiva legata all'X. Le donne possono ereditare la malattia, ma a causa della loro doppia struttura cromosomica X, raramente si ammalano. Gli uomini malati non sono in grado di riprodursi.
Sebbene la mutazione sia genetica, oltre a questo fattore interno, probabilmente fattori esterni giocano un ruolo nell'insorgenza della malattia. A causa della rarità e della base di ricerca limitata, il ruolo di questi fattori esterni non è stato ancora chiarito in modo definitivo.
Puoi trovare i tuoi farmaci qui
➔ Medicinali per la debolezza muscolareSintomi, disturbi e segni
La sindrome di Allan-Herndon-Dudley è una malattia congenita che di solito si manifesta nei bambini piccoli o nei neonati. Le persone colpite soffrono di debolezza muscolare più o meno grave. Il tessuto muscolare dei bambini è notevolmente sottosviluppato. La debolezza dei muscoli è presto accompagnata da deformità articolari.
Anche le contratture sono sintomi di accompagnamento frequenti. La mobilità dei bambini è sempre più compromessa dalle contratture e dalle deformità. Per questo motivo, le persone colpite spesso appaiono innaturalmente statiche o addirittura immobili. A causa della scarsa fornitura di ormoni tiroidei correlata alla mutazione, le persone colpite spesso soffrono anche di crampi muscolari o compiono movimenti involontari con le braccia e le gambe.
Spesso le persone colpite non possono muoversi in modo indipendente. Nella maggior parte dei casi, le menomazioni motorie sono associate a gravi disturbi mentali. Ad esempio, la maggior parte dei pazienti non è in grado di parlare. In singoli casi, l'AHDS può essere caratterizzato da molti altri sintomi nell'area dello sviluppo mentale e fisico.
Diagnosi e corso
Il medico di solito sospetta prima l'AHDS nell'anamnesi. Nei test di laboratorio, un aumento del livello di T3 con normali livelli di FT4 e TSH indica la sindrome di Allan-Herndon-Dudley. L'imaging del sistema nervoso centrale fa solitamente parte della diagnosi.
Le debolezze muscolari dovute a malattie dei motoneuroni possono essere escluse dalla diagnosi differenziale. La prognosi per i pazienti con sindrome di Allan-Herndon-Dudley è relativamente scarsa. Finora la malattia è incurabile. Gli studi hanno suggerito che la tempistica della diagnosi può svolgere un ruolo critico nella prognosi del paziente.
complicazioni
Come tutte le malattie ereditarie cromosonali, la sindrome di Allan - Herndon - Dudley non può essere trattata in modo curativo. L'effetto collaterale più comune della sindrome di Allan - Herndon - Dudley - pronunciata debolezza muscolare - può essere trattato con la fisioterapia. Tale trattamento volto a rafforzare i muscoli può essere doloroso per il paziente.
I bambini piccoli, in particolare, spesso rifiutano la terapia a causa del dolore. Nonostante l'allenamento intensivo, la fisioterapia non sempre porta al successo desiderato. È simile al supporto logopedico del paziente Allan - Herndon - Dudley. Sebbene la riduzione della capacità linguistica possa essere migliorata con un allenamento intensivo, il trattamento non sempre porta al successo a causa della disabilità mentale per lo più di alto grado delle persone colpite.
La frustrazione da parte del paziente e lo stress su tutta la famiglia sono una delle complicazioni più gravi nel trattamento della sindrome di Allan - Herndon - Dudley. Crampi muscolari e movimenti delle estremità che non possono essere influenzati possono essere trattati con la somministrazione di miorilassanti. Le complicazioni possono essere viste negli effetti collaterali a volte gravi dei farmaci.
Oltre allo stress a carico del tratto gastrointestinale sono da segnalare la stanchezza, una sensazione generale di spossatezza e malessere. L'uso a lungo termine di relanxants danneggia anche il fegato e i reni. Se la sindrome di Allan - Herndon - Dudley non viene trattata, le persone colpite non saranno in grado di compiere progressi significativi in termini di capacità mentali o motorie.
Quando dovresti andare dal dottore?
In molti casi, il trattamento diretto della sindrome di Allan-Herndon-Dudley non è possibile. Per questo motivo il trattamento è prevalentemente sintomatico ed è rivolto ai singoli reclami e ritardi. Di norma, i genitori dovrebbero quindi consultare un medico se il bambino ha debolezza muscolare.
Questo può diventare evidente per esaurimento o stanchezza persistente. La consulenza medica è necessaria anche se la sindrome di Allan-Herndon-Dudley rallenta lo sviluppo mentale e motorio.
Se il trattamento non avviene durante l'infanzia, può portare a notevoli disagi e limitazioni nell'età adulta. Un medico dovrebbe essere consultato, soprattutto se il paziente non può più parlare. Il trattamento è necessario anche per i crampi muscolari. Se si tratta di un'emergenza acuta, puoi andare direttamente in ospedale o chiamare un'ambulanza.
Nella maggior parte dei casi, la sindrome di Allan-Herndon-Dudley sarà trattata da un medico generico o da un pediatra. Tuttavia, i singoli reclami devono essere esaminati e trattati dal rispettivo specialista o terapista.
Medici e terapisti nella tua zona
Trattamento e terapia
L'AHDS è una malattia causalmente non curabile. Poiché non sono disponibili terapie per correggere la causa primaria, la malattia non è stata finora curabile. Nel frattempo, i progressi nel campo della terapia genica suggeriscono che gli approcci di terapia genica saranno presto approvati per la pratica clinica quotidiana.
Fino a che punto i pazienti con la sindrome trarrebbero beneficio dall'approvazione non è stato ancora chiarito. Al momento non esiste un'opzione di trattamento stabilita o standardizzata per i pazienti con AHDS nemmeno nel campo della terapia sintomatica. Alcuni anni fa i ricercatori consideravano la somministrazione di TRIAC una possibile opzione di terapia sintomatica.
TRIAC è un ormone tiroideo non classico, il triiodotiroacetato. La somministrazione dell'ormone è stata effettuata in uno studio clinico su bambini affetti, ma non è stato possibile ottenere risultati visibili. I risultati dello studio non sono necessariamente significativi perché la somministrazione dell'ormone è iniziata relativamente tardi.
Per questo motivo, nel 2014 TRIAC era ancora considerata la migliore terapia possibile. In un caso nel 2014, è stato documentato un significativo miglioramento dello sviluppo motorio e mentale durante il trattamento con TRIAC. La terapia è stata avviata sulla persona affetta nella prima infanzia.
I risultati degli studi fino ad oggi indicano quindi che l'ora in cui viene iniziata la terapia ha un effetto sui risultati della terapia che non deve essere sottovalutato per i pazienti con AHDS. Terapie di supporto di accompagnamento come la terapia occupazionale e la fisioterapia o l'intervento precoce possono teoricamente essere utilizzate per migliorare la qualità della vita e le capacità dei pazienti. Tuttavia, non vi è quasi alcuna prova dell'efficacia di un tale approccio in relazione ai pazienti con AHDS.
Outlook e previsioni
La sindrome di Allan-Herndon-Dudley causa una serie di sintomi diversi nella maggior parte dei pazienti. Innanzitutto, le persone colpite soffrono di grave debolezza muscolare. Ciò significa che le normali attività o sport non possono più essere svolte facilmente per la persona interessata. Ci sono anche notevoli ritardi nello sviluppo intellettuale e mobile. La concentrazione del paziente è chiaramente limitata e ridotta.
Ci sono ancora forti crampi nei muscoli e quindi spesso movimenti involontari o spasmi. Con il progredire della sindrome di Allan - Herndon - Dudley, le persone colpite non possono più parlare. La vita quotidiana del paziente è notevolmente limitata dalla sindrome e la qualità della vita è ridotta. In alcuni casi, i pazienti dipendono quindi dall'aiuto di altre persone nella loro vita quotidiana.
Di solito non è possibile trattare la sindrome di Allan-Herndon-Dudley in modo causale. Per questo motivo il trattamento è solo sintomatico. Le persone colpite dipendono da varie terapie, che però non sempre portano a un decorso positivo della malattia. In alcuni casi, la sindrome di Allan-Herndon-Dudley limita l'aspettativa di vita delle persone colpite.
Puoi trovare i tuoi farmaci qui
➔ Medicinali per la debolezza muscolareprevenzione
L'AHDS può essere prevenuto solo attraverso la consulenza genetica. I portatori di mutazione possono, ad esempio, decidere di non avere figli propri.
Dopo cura
La necessità di cure di follow-up per la sindrome di Allan-Herndon-Dudley di origine genetica colpisce solo i neonati maschi. Il problema è che non esiste una forma di terapia adeguata per questa condizione ereditaria. Le gravi conseguenze dei difetti nei trasmettitori degli ormoni tiroidei difficilmente possono essere migliorate. I tentativi di fornire sollievo ai bambini colpiti dando loro speciali ormoni tiroidei sono falliti.
Il problema è che la base della malattia di solito è già stabilita nel corpo della madre. Danneggiano permanentemente il nascituro. Da questo punto di vista il trattamento inizia troppo tardi, cioè solo dopo il parto. Nell'aftercare, solo il danno che è già presente può essere trattato. Tuttavia, c'è speranza. Nel 2014 è diventato noto un caso in cui un bambino con sindrome di Allan-Herndon-Dudley è stato trattato con successo con TRIAC. Il follow-up era ancora necessario perché il bambino non poteva essere curato. Almeno i suoi sintomi sono stati alleviati.
La sindrome di Allan-Herndon-Dudley è in parte causata da una barriera ematoencefalica difettosa. Questo è ciò che suggeriscono gli studi al Cedars Sinai Hospital. Gli ormoni tiroidei non possono funzionare a causa della barriera ematoencefalica difettosa. Questo vale anche per gli ormoni tiroidei che vengono somministrati dopo la nascita del bambino. La biotecnologia o la ricerca genetica possono essere d'aiuto. Tutti i tentativi di trattamento stanno attualmente fallendo. Ciò influisce anche sull'assistenza successiva ai bambini gravemente danneggiati.
Puoi farlo da solo
La sindrome di Allan-Herndon-Dudley è una condizione grave che non è stata ancora trattata in modo efficace. Tuttavia, i genitori possono ancora intraprendere alcune azioni per supportare la terapia.
Prima di tutto, è importante un regolare allenamento cognitivo e attività fisica. Una terapia completa, che, a seconda della gravità della sindrome, può essere composta da esercizi di linguaggio e lettura, ma anche di esercizi cerebrali generali, offre anche esercizi di fisioterapia. La formazione deve essere adattata individualmente ai sintomi. I genitori dei bambini colpiti dovrebbero quindi assicurarsi che le misure siano scelte in modo ottimale e che il bambino non sia sopraffatto.
In caso di gravi disturbi mentali, il bambino potrebbe aver bisogno di un sostegno permanente nella vita di tutti i giorni. Un servizio di assistenza ambulatoriale può essere un importante sollievo per i genitori. Altrettanto importante è il trattamento ospedaliero, che può essere supportato a casa da un monitoraggio regolare dei sintomi.
I genitori dovrebbero anche avvalersi della consulenza psicologica e, se necessario, anche rivolgersi a un gruppo di auto-aiuto, perché il contatto con altre persone affette rende più facile affrontare la malattia. Inoltre, i genitori spesso ricevono importanti suggerimenti su come affrontare un bambino malato.


.jpg)

.jpg)

.jpg)

.jpg)



.jpg)


.jpg)