Al Sindrome di Apert è una rara malattia ereditaria. Le malformazioni si verificano in tutto l'organismo con le perdite funzionali croniche più gravi e il decorso della malattia attraverso le malformazioni. Entrambi i sessi sono ugualmente colpiti.
Cos'è la sindrome di Apert?

© Sashkin - stock.adobe.com
La sindrome di Apert, nota anche come Sindrome da acrocefalosindattilia noto per essere la causa di malformazioni da gravi a estremamente gravi che possono interessare tutto il corpo. Queste molteplici malformazioni furono descritte per la prima volta dal pediatra francese Eugène Apert, da cui prese il nome anche la malattia incurabile. Eugène Apert esercitò come pediatra nella capitale francese Parigi e visse dal 1868 al 1940.
La sindrome di Apert è solo una delle possibili altre manifestazioni fenotipiche della sindrome da acrocefalosindattilia. Nel registro della malattia dell'ICD-10, la sindrome di Apert ha il codice di diagnosi Q87.0. Le altre 4 malattie, che fanno anche parte delle sindromi acrocefalosindattilia, includono la sindrome di Crouzon, la sindrome di Carpenter, la sindrome di Pfeiffer e la sindrome di Saethre-Chotzen. La sindrome di Pfeiffer non deve essere confusa con la mononucleosi infettiva, nota anche come malattia di Pfeiffer.
Tra le malattie delle sindromi da acrocefalosindattilia citate, invece, la sindrome di Apert è quella con il decorso più grave e fulminante. Una volta effettuata la diagnosi sospetta, i medici si trovano ad affrontare la sfida di assegnare il quadro clinico alla corretta diagnosi da parte del gruppo delle sindromi da acrocefalosindattilia.
cause
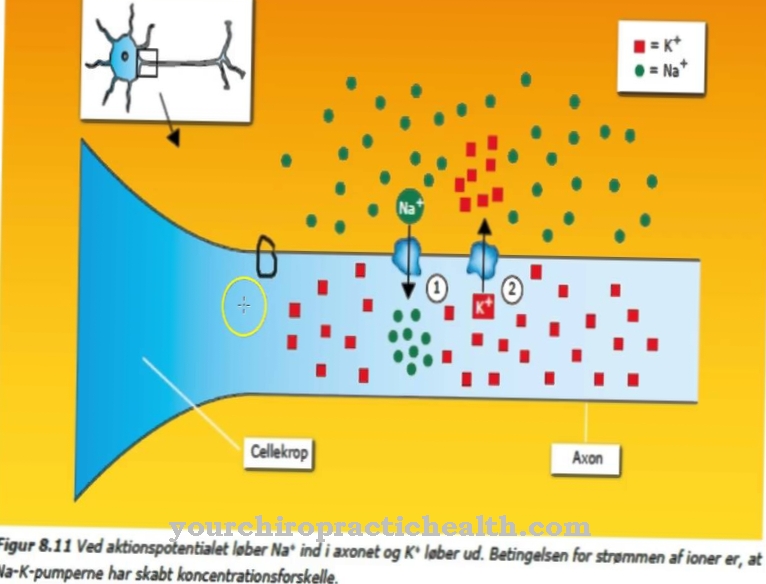
Tutte le sindromi da acrocefalosindattilia, inclusa la sindrome di Apert, si basano su una speciale mutazione genetica. I test biologici molecolari hanno nel frattempo stabilito che il fenotipo della sindrome di Apert, cioè l'aspetto esteriore visibile della malattia, può essere fatto risalire a una mutazione del decimo cromosoma. All'epoca, tuttavia, queste connessioni non erano ancora note allo scopritore della malattia. Sul cromosoma 10 si trova il modello per il cosiddetto gene del recettore del fattore di crescita dei fibroblasti, FGFR.
Il locus genetico esatto è 10Q26, la sindrome di Apert viene ereditata tramite quella che è nota come eredità autosomica dominante. Se il gene corrispondente è mutato sul cromosoma 10, allora c'è sempre un focolaio fenotipico della malattia, in questo contesto si parla di penetranza completa. La varianza nella sindrome di Apert è molto alta; questo è di rilevanza clinica in quanto i segni ei sintomi della malattia possono essere molto diversi nelle persone colpite. Le cause più comuni della sindrome di Apert sono spontanee, ovvero mutazioni imprevedibili sul decimo cromosoma.
Sintomi, disturbi e segni
Le indagini di biologia molecolare hanno dimostrato oltre ogni dubbio che l'età del padre del bambino gioca un ruolo importante nella tendenza alla mutazione spontanea. La sindrome di Apert si manifesta quindi in particolare nei figli di padri anziani. Questa relazione è stata verificata per la sindrome di Apert, ma non per le altre sindromi da acrocefalosindattilia. Statisticamente, un neonato su 130.000 è affetto dalla sindrome di Apert. I sintomi, i reclami ei segni che possono indicare la presenza di una sindrome di Apert sono estremamente diversi e variabili.
Nel caso di un quadro clinico completamente sviluppato, la diagnosi corretta viene effettuata in modo affidabile, con sottomaschere, tuttavia, può esserci anche un ritardo nella diagnosi corretta. I sintomi cardinali della sindrome di Apert comprendono malformazioni del cranio osseo, del viso, delle estremità comprese mani e piedi, nonché scoliosi della colonna vertebrale, problemi respiratori, ametropia e perdita dell'udito.
In oltre l'80% dei casi, il medico può anche determinare ciò che è noto come idrocefalo e grave ritardo mentale. Le ossa del cranio sono così fuse insieme che si verificano contusioni del cervello e quindi un aumento della pressione intracranica.
Diagnosi e corso
Malformazioni e caratteristiche tipiche di una sindrome di Apert possono essere riconosciute con un'ecografia fine intorno al quarto mese di gravidanza. Immediatamente dopo la nascita, si può fare una prima diagnosi di sospetto sulla base dei tipici sintomi indicativi. La diagnosi deve essere confermata e confermata immediatamente da ulteriori esami del sangue genetico molecolare. Se la diagnosi viene quindi stabilita oltre ogni dubbio, la terapia chirurgica deve essere avviata immediatamente.
complicazioni
Nella sindrome di Apert ci sono complicazioni molto gravi che influenzano in modo grave la vita del paziente. La sindrome di Apert provoca gravi malformazioni sul corpo che possono portare a problemi di salute. Sia le donne che gli uomini sono affetti dalla sindrome.
Di regola, la sindrome si presenta come una malformazione nell'osso del cranio. Il cranio quindi ha un aspetto diverso rispetto alle persone sane. Ciò porta spesso a bullismo e prese in giro, soprattutto nei bambini, e può innescare problemi psicologici nei pazienti. Le estremità possono anche contenere malformazioni e ci sono anche problemi respiratori e perdita dell'udito.
La maggior parte dei malati ha bisogno di un aiuto visivo. Oltre ai problemi fisici e fisici, la maggior parte dei pazienti sperimenta anche problemi mentali e psicologici. Il pensiero e l'azione sono severamente limitati, cosicché nella maggior parte dei casi il paziente non può far fronte da solo alla vita quotidiana e dipende anche dall'aiuto della famiglia.
Il trattamento è possibile solo in misura limitata. I sintomi fisici possono essere trattati con interventi chirurgici in modo da rimuovere deformità e disallineamenti. Il ritardo mentale non può essere trattato, tuttavia, in modo che i pazienti abbiano anche un'aspettativa di vita più breve.
Quando dovresti andare dal dottore?
Nella sindrome di Apert, la diagnosi precoce e il trattamento della malattia sono di grande importanza. Per questo motivo, un medico dovrebbe essere consultato ogni volta che si verificano i sintomi di questa sindrome. Di norma, le persone colpite soffrono di varie deformità del corpo, che tuttavia non devono verificarsi immediatamente dopo la nascita. Pertanto, se il paziente ha difficoltà a respirare o alla vista, deve essere eseguita una visita medica. Un esame e un trattamento da parte di un medico sono utili anche in caso di perdita dell'udito che si manifesta senza un motivo particolare.
Non è raro che i pazienti soffrano di un aumento del ritardo dovuto alla sindrome di Apert. Questo può manifestarsi attraverso difficoltà a scuola e disturbi mentali e motori. Se si verificano anche questi sintomi, è molto probabile che il paziente abbia la sindrome di Apert.
Con il trattamento precoce, i sintomi possono essere limitati in modo che la vita di tutti i giorni sia sopportabile per la persona interessata. Molti sintomi possono essere risolti o alleviati attraverso vari interventi chirurgici. Una visita dal medico può essere utile anche ai genitori o ai parenti del bambino se soffrono di disturbi psicologici o depressione.
Medici e terapisti nella tua zona
Trattamento e terapia
L'obiettivo della terapia è rimuovere le deformità del cranio responsabili dell'aumento della pressione intracranica con l'idrocefalo. L'intervento terapeutico necessario per questo può essere eseguito solo chirurgicamente. Per fare questo, le suture craniche, le suture, devono essere sabbiate in anestesia generale. Questa speciale forma di intervento chirurgico può essere eseguita solo in centri qualificati.
Il disallineamento delle dita dei piedi e delle dita viene solitamente eseguito in modo sincrono, per quanto possibile. In un'altra procedura separata, le strutture dell'orecchio medio devono essere operate perché i tipici ossicini, martello, incudine e staffa non sono o non sono completamente sviluppati nella sindrome di Apert.
In alcuni casi, questo intervento chirurgico può aiutare i bambini a recuperare almeno una parte dell'udito. Sfortunatamente, se le deformità dell'orecchio medio sono troppo pronunciate, l'udito non può essere ripristinato. I bambini colpiti sono esposti a uno stress psicologico molto estremo, perché il rifiuto dall'ambiente è molto alto. E i bambini colpiti sono già sottoposti a un gran numero di interventi nei primi anni di vita. Anche le limitazioni psicologiche che sono aggravate di conseguenza dovrebbero essere sempre trattate.
Outlook e previsioni
La prognosi per la sindrome di Apert deve essere classificata come sfavorevole. La malattia ereditaria si verifica più frequentemente nei bambini con un padre anziano ed è ancora considerata incurabile fino ad oggi. Per motivi legali, a medici e scienziati è vietato interferire con la genetica umana. Pertanto, con le opzioni attualmente disponibili, non esiste una terapia o un farmaco che possa portare a una guarigione dalla malattia.
Il piano di trattamento ha lo scopo di alleviare il disagio esistente e si concentra sul miglioramento della qualità della vita. In molti casi, tuttavia, il trattamento non può essere eseguito poiché non vedrebbe alcun ulteriore successo. Sebbene la malattia sia incurabile, non è progressiva. Le menomazioni esistenti non peggiorano nel corso della vita.
Gli ausili per la vista vengono spesso utilizzati durante il trattamento per migliorare la vista. Inoltre, se possibile, viene eseguito un intervento chirurgico per modificare le deformità del cranio. Questo non ha solo un carattere cosmetico, ma serve anche a garantire che vasi e tessuti non vengano schiacciati o feriti durante il processo di crescita. Queste sono spesso misure preventive in modo che non possano verificarsi insufficienza d'organo acuta o sanguinamento. Ciò comporterebbe la morte prematura del paziente.
prevenzione
La prevenzione diretta delle mutazioni spontanee non è ancora possibile. Tuttavia, la sindrome da acrocefalosindattilia può essere rilevata precocemente in una cosiddetta diagnosi prenatale. Dalla nona settimana di gravidanza è possibile anche un test di amniocentesi, con il quale si può già fare la diagnosi. Se è probabile la sindrome da acrocefalosindattilia, i medici di solito le consigliano di interrompere la gravidanza il prima possibile.
Dopo cura
La sindrome di Apert è sempre associata a una terapia lunga. I malati necessitano di supporto medico e terapeutico per mesi o anni, poiché i vari disturbi e sintomi possono ripresentarsi o persistere in forma cronica. Di conseguenza, l'assistenza di follow-up si concentra su controlli di follow-up regolari.
I pazienti dovrebbero consultare un medico settimanalmente all'inizio. Se la malattia progredisce positivamente, i controlli possono essere gradualmente ridotti, per cui gli esami di routine devono essere eseguiti in modo permanente. Se insorgono complicazioni durante il follow-up, il trattamento deve essere ripreso.
I pazienti dovrebbero anche cercare i farmaci di cui hanno bisogno per alleviare efficacemente le infezioni del seno, le difficoltà respiratorie e altri sintomi, a volte cronici. Aftercare include anche il cambiamento di alcune abitudini di vita, che possono esacerbare i sintomi. Quindi la dieta deve essere cambiata.
Una dieta povera di grassi e ricca di vitamine può ridurre l'infiammazione e sostenere il sistema immunitario. A causa dei diversi percorsi che la malattia genetica può intraprendere, i passaggi esatti devono essere sempre elaborati tenendo conto del profilo sintomatico individuale del paziente. Il medico responsabile è la persona di contatto giusta per l'assistenza di follow-up dopo la sindrome di Apert.
Puoi farlo da solo
La sindrome di Apert è una malattia grave associata a numerosi disturbi e un notevole stress fisico e psicologico per le persone colpite. Le persone colpite dovrebbero quindi avvalersi di misure terapeutiche che supportano il trattamento medico convenzionale.
Lo scambio con altre persone colpite è possibile come parte di un gruppo di auto-aiuto. Questo non solo ti consente di scambiare consigli su come affrontare la malattia, ma anche di stabilire nuovi contatti con altri malati e specialisti. A lungo termine, tuttavia, la malattia deve essere accettata, dalle persone colpite e dai loro parenti allo stesso tempo. L'iniziativa dei genitori Apert Syndrome and Related Malformations e.V. offre alle persone colpite ulteriori opzioni di contatto e suggerimenti per affrontare la malattia in modo sano.
Oltre a queste misure terapeutiche, i sintomi possono essere alleviati anche da uno stile di vita sano. L'esercizio fisico regolare e una dieta sana aiutano a lungo termine a contenere i sintomi e migliorare la qualità della vita. Inoltre, dovrebbero essere prese le misure necessarie per rendere la vita futura accessibile ai disabili. Le misure menzionate sono meglio implementate in consultazione con il medico responsabile e i parenti.
.jpg)

.jpg)

.jpg)



.jpg)