L'ipossantina è una purina deviata e si presenta in forma legata come basi azotate e in forma libera, ad es. B. nelle urine. Si trova anche nelle ghiandole e nel midollo osseo. Come prodotto di deaminazione dell'adenina, l'ipoxantina viene ossidata in acido urico e xantina. Meno spesso forma una struttura di base di acidi nucleici.
Che cos'è l'ipoxantina guanina fosforibosiltransferasi?
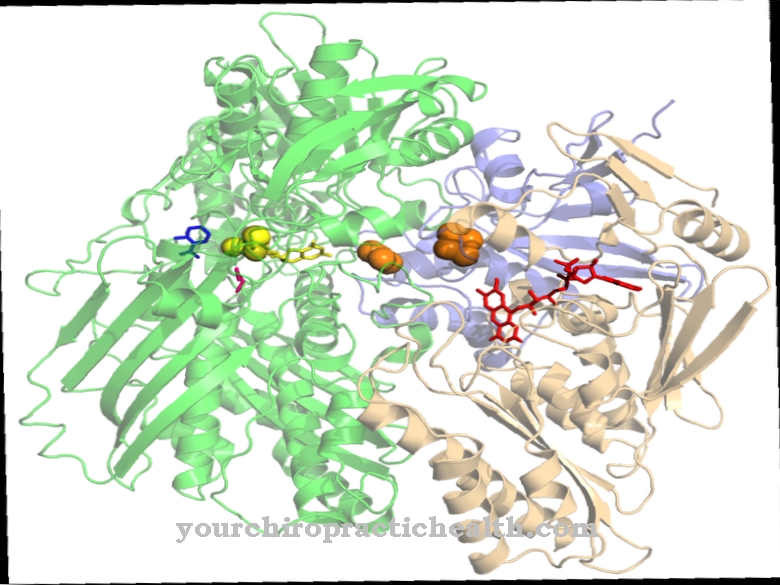
L'enzima tetramerica è formato da ipoxantina e guanina Ipoxantina guanina fosforibosil transferasi.
I tetrameri sono macromolecole costituite da quattro elementi costitutivi simili, più precisamente da monomeri. L'enzima è uno dei più importanti nel metabolismo delle purine degli eucarioti, è sensibile ai cambiamenti genetici e può causare deviazioni nell'uomo attraverso mutazioni geniche, che si esprimono in alcune malattie metaboliche. Tali sono z. B. Sindromi di Lesch-Nyhan e Kelley-Seegmiller.
Funzione, effetto e compiti
L'enzima ipoxantina-guanina-fosforibosiltransferasi aumenta il metabolismo delle purine e la sua efficacia energetica.
Questo si basa su basi puriniche, che sono acidi nucleici strutturalmente derivati dalle purine. Questi sono xantina, ipoxantina, adenina e guanina, che si attaccano ad altre basi attraverso legami idrogeno. Tali legami hanno una grande influenza sulla doppia elica del DNA e sulla replicazione e svolgono un ruolo nella biosintesi delle proteine.
Le basi purine possono essere riciclate da due enzimi. Oltre alla ipoxantina guanina fosforibosil transferasi, questa è l'adenina fosforibosil transferasi. Entrambi formano un nucleotide attraverso un residuo fosforibosilico, che a sua volta è un elemento costitutivo di base degli acidi nucleici sia nel DNA che nell'RNA. La molecola è composta da zucchero, base e fosfato e controlla le funzioni regolatorie vitali nelle cellule. Durante l'accumulo, l'ATP viene salvato e la formazione di acido urico viene ridotta.
Se le basi purine vengono riciclate, viene indicato come percorso di salvataggio. Questo è un termine generale per le vie metaboliche in cui una sintesi di biomolecole deriva da prodotti di degradazione. L'organismo esegue il proprio processo di riciclaggio, in base al quale circa il novanta per cento delle basi puriniche viene riutilizzato e il dieci per cento viene effettivamente escreto. Ciò mostra l'efficienza del riciclaggio delle basi puriniche e l'importanza dell'ipoxantina-guanina fosforibosil transferasi.
Istruzione, occorrenza, proprietà e valori ottimali
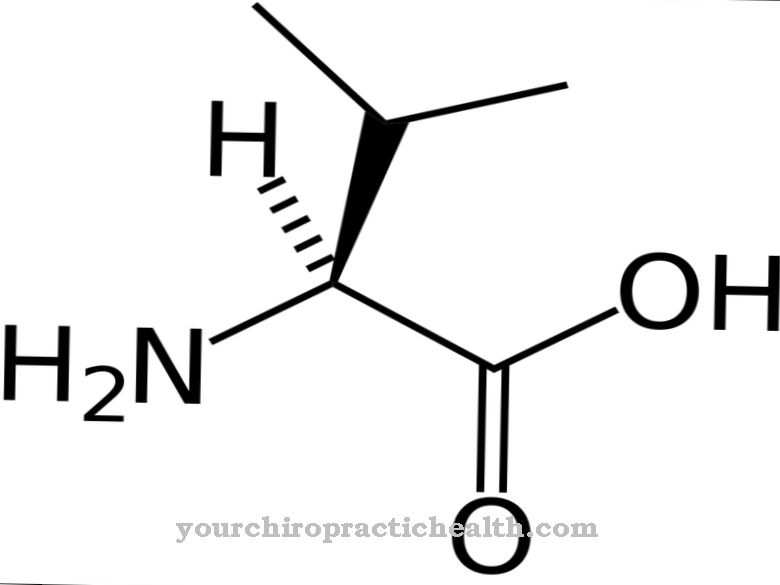
Se si verificano mutazioni nel gene HPRT, le dimensioni e gli amminoacidi possono cambiare. Questa può essere l'incorporazione di ulteriori sequenze di DNA o nucleotidi, che a sua volta porta a una produzione errata del prodotto genico che è codificato sul rispettivo gene, o anche alla delezione dell'intera sequenza. È z. B. cambia la sequenza degli amminoacidi, insorgono malattie come la gotta.
Le malattie metaboliche come la sindrome di Lesch-Nyhan a seguito di un difetto genetico sono particolarmente gravi. Questo è ereditato in modo recessivo legato all'X, il che significa che colpisce principalmente gli uomini che hanno un solo cromosoma X. Il difetto genetico può essere presente nelle donne, ma si manifesta come malattia solo quando entrambi i cromosomi X sono colpiti, il che è relativamente raro. Molto spesso il secondo cromosoma X compensa il difetto del primo.
Puoi trovare i tuoi farmaci qui
➔ Medicinali per la salute della vescica e delle vie urinarieMalattie e disturbi
La sindrome si manifesta in una carenza dell'ipoxantina guanina fosforibosil transferasi. L'enzima non viene prodotto a causa del difetto genetico. A causa della mutazione e della mancanza di riciclaggio e conversione delle basi guanina e ipoxantina, si verifica un accumulo delle basi puriniche che devono essere accumulate ed escrete dall'organismo.
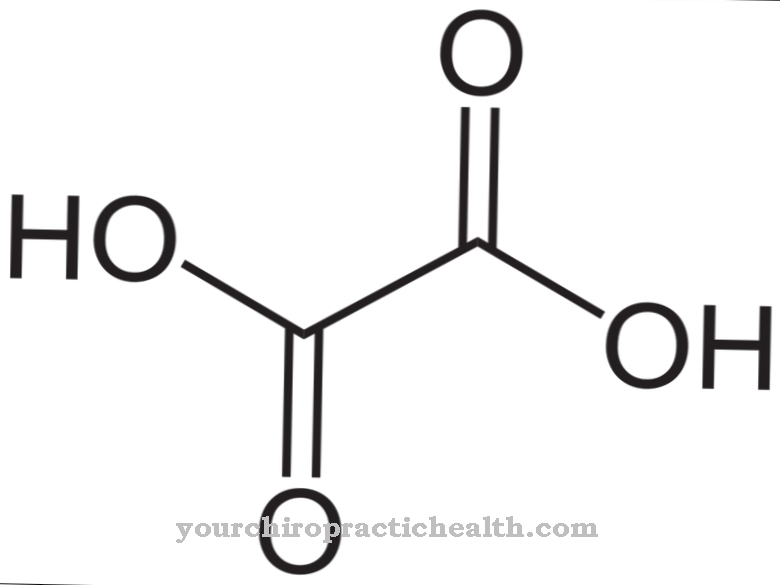
La degradazione avviene tramite il prodotto intermedio xantina, che viene convertito in acido urico ed escreto attraverso i reni. Se questo processo è limitato, i cristalli di acido urico si formano nell'area delle articolazioni, che quindi innescano più attacchi di gotta. L'enzima non viene più prodotto, il livello di acido urico nei tessuti e nel sangue aumenta e il sistema nervoso centrale è disturbato.
La sindrome di Lesch-Nyhan non è direttamente visibile alla nascita. Una notevole posizione delle gambe e la tendenza del bambino a muoversi poco e svilupparsi più lentamente possono essere viste solo dopo una decina di mesi. La sindrome è debole e grave. Una maggiore secrezione di acido urico e attacchi di gotta più leggeri sono la forma più lieve; se sono gravi, si verificano autolesionismo, grave compromissione mentale e aggressività. L'autolesionismo si verifica attraverso morsi di dita o labbra. Quando si mordono le estremità, si può spesso osservare che le persone colpite limitano la loro autoaggressione a una sola mano. L'aggressione, a sua volta, è spesso diretta contro le persone a te vicine come fratelli o genitori.
La forma più grave della malattia è caratterizzata da molteplici disfunzioni neurologiche e da una tendenza molto pronunciata all'automutilazione. La sindrome si manifesta in spasticità, distonia, ipotonia, coreoatetosi e una maggiore disponibilità a reagire ai riflessi. Le qualità mentali e lo sviluppo sono gravemente limitati. In questa condizione, la sindrome può anche portare alla morte in misura particolarmente drastica.
La malattia viene diagnosticata attraverso un quadro medico. Viene misurato il livello di acido urico nelle urine e nel sangue e l'attività dell'ipoxantina-guanina fosforibosiltransferasi nei tessuti e nel sangue. Quest'ultimo è notevolmente ridotto e può essere presente anche prenatale.
Il trattamento della malattia è difficile. La cura non è possibile e senza cure il bambino muore nei primi anni di vita. In alcuni casi, i denti da latte devono essere estratti come misura preventiva. Altri approcci terapeutici includono l'abbassamento dei livelli di acido urico utilizzando farmaci come l'allopurinolo, che agisce come un inibitore della gotta. Le basi puriniche non vengono riciclate, ma l'acido urico viene scomposto meglio. Vengono trattati anche i rispettivi disturbi, infezioni e danni ai nervi e si consiglia una dieta speciale, che di solito non contiene carne ed è povera di purine.
Sono inoltre in corso ricerche sugli effetti collaterali psicosomatici della stimolazione cerebrale profonda. La medicina spera che questo impedisca l'aggressione e l'automutilazione. La sindrome di Kelley-Seegmiller, d'altra parte, è la forma più lieve di carenza di ipoxantina-guanina fosforibosiltransferasi. Anche qui si produce troppo acido urico e si manifestano le prime malattie della gotta. Le prime indicazioni della sindrome sono cristalli arancioni nel pannolino del bambino, infezioni del tratto urinario e urolitiasi. La gotta o l'artrite acuta si sviluppano durante la pubertà.
Un sottosviluppo mentale e auto-attacchi, come si verificano con la sindrome di Lesch-Nyhan, non sono il caso, al massimo possono portare a disturbi dell'attenzione. Il trattamento precoce di solito consente alle persone colpite di avere un'aspettativa di vita normale.
.jpg)

.jpg)

.jpg)



.jpg)


.jpg)