Il Distrofia Miotonica, tipo 1 (sindrome di Curschmann-Steinert) è una malattia multisistemica ereditaria autosomica dominante con i principali sintomi di debolezza muscolare e opacità del cristallino (cataratta). Si distingue tra due forme di malattia: una forma congenita (congenita), in cui il neonato è evidente a causa della debolezza muscolare ("infante floscia"), e una forma adulta, che si manifesta solo nella 2a-3a decade di vita. La Distrofia Miotonica di tipo 1 è incurabile e, a seconda della sua gravità e progressione, riduce l'aspettativa di vita.
Cos'è la Distrofia Miotonica di tipo 1?
© peshkova - stock.adobe.com
Il Distrofia miotonica di tipo 1 è una delle cosiddette malattie da ripetizione trinucleotidica. Nel codice genetico sul braccio lungo del cromosoma 19, viene duplicato un trinucleotide dalle basi azotate citosina, timina e guanina.
Mentre questa tripletta di base viene ripetuta 5-35 volte nelle persone sane, in quelle con sintomi lievi è di circa 50-200, nelle forme gravi anche oltre 1000 ripetizioni. Il trinucleotide non codifica direttamente una proteina, ma influenza la sintesi di altre proteine. Un enzima richiesto nei muscoli scheletrici e cardiaci, la proteina chinasi della distrofia miotonica (DMPK), viene prodotto in modo ridotto a causa del difetto genetico.
Ma anche altre proteine sono interessate, ad es. il SIX5 espresso nel cristallino o nel recettore dell'insulina. Pertanto, la distrofia miotonica di tipo 1 colpisce molti sistemi di organi diversi. Con un'incidenza di circa 1: 20.000, la Distrofia Miotonica di tipo 1 è la più comune miotonia e allo stesso tempo la più comune distrofia muscolare che si manifesta in età adulta.
In termini di ereditarietà, il numero di ripetizioni di trinucleotidi aumenta di generazione in generazione, in modo che l'insorgenza della malattia nella prole sia precoce e più grave. La forma congenita viene sempre ereditata dalla madre. La distrofia miotonica di tipo 1 colpisce i ragazzi più frequentemente rispetto alle ragazze.
cause
Nella forma congenita, il bambino si nota subito dopo la nascita con debolezza muscolare generalizzata, labbro superiore rialzato e insufficienza respiratoria. A causa dei problemi respiratori, molti neonati dipendono dalla respirazione artificiale e il 25-50% muore entro i primi 18 mesi di vita.
Nei bambini che sopravvivono più a lungo sono prevedibili ritardi nello sviluppo e grave ritardo mentale. La loro aspettativa di vita è di circa 30-40 anni. Se la malattia si manifesta solo in età adulta, le persone colpite spesso notano per prima cosa debolezza muscolare nei muscoli distanti dal busto, specialmente nelle gambe, nel collo e nella zona del viso. I muscoli facciali si atrofizzano, rendendo il paziente magro.
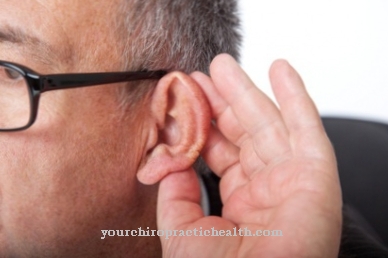
Ne derivano anche disturbi della parola e della deglutizione. La debolezza muscolare è accompagnata da un rilassamento muscolare ritardato, per cui è difficile per le persone colpite, ad esempio, allentare nuovamente un movimento. Ulteriori sintomi sono opacità del cristallino, perdita dell'udito dell'orecchio interno, aumento del bisogno di sonno, limitazioni cognitive e ridotta tolleranza al glucosio fino al diabete mellito. A causa del disturbo dell'equilibrio ormonale, l'atrofia testicolare e la calvizie si verificano tipicamente negli uomini e disturbi mestruali nelle donne.
Gli effetti sui muscoli cardiaci sono particolarmente pericolosi: si verificano spesso aritmie cardiache e occasionalmente anche arresto cardiaco. Se la debolezza muscolare raggiunge i muscoli centrali, ne risultano disturbi respiratori e una maggiore suscettibilità alle infezioni dei polmoni. La distrofia miotonica di tipo 1 è sempre progressiva, ma la gravità e la composizione dei suoi sintomi sono estremamente variabili. In media, l'aspettativa di vita nella forma adulta di Distrofia Miotonica di tipo 1 è di circa 50-60 anni.
Sintomi, disturbi e segni
La caratteristica principale della distrofia miotonica di tipo 1 è il rilassamento ritardato dei muscoli dopo una contrazione muscolare. Questa caratteristica può essere utilizzata per distinguere la malattia da altre distrofie muscolari. I muscoli più lontani dal tronco, come viso, collo, avambraccio, mano, parte inferiore della gamba e muscoli del piede, sono particolarmente colpiti. Ci sono altri sintomi indipendenti dai disturbi muscolari.
Spesso si verificano aritmie cardiache o insufficienza cardiaca. A causa del coinvolgimento del cuore, gli incidenti anestetici si verificano spesso durante l'anestesia. Si osservano spesso cataratta e calvizie negli uomini. Il livello di testosterone è troppo basso, il che spesso porta alla perdita di testicoli. C'è un aumento del rischio di sviluppare il diabete.
Parlare e deglutire sono difficili per il paziente. Inoltre, il paziente è costantemente stanco durante il giorno, il che può portare a pause respiratorie durante la notte. Tuttavia, l'apnea notturna non è sempre presente. Possono verificarsi anche disturbi digestivi, disturbi della cistifellea o disturbi dell'udito come ulteriori sintomi. Sebbene sia una condizione ereditaria, i sintomi non compaiono in molti pazienti fino a quando non hanno 20 anni.
Una cataratta viene spesso diagnosticata come primo segno della malattia. Tuttavia, esiste anche una forma della malattia che esiste dalla nascita. Questa forma congenita di miotonia muscolare è caratterizzata da un decorso particolarmente grave con insufficienza respiratoria pericolosa per la vita e disturbi dello sviluppo mentale e fisico.
Diagnosi e corso
Se sospetti Distrofia miotonica di tipo 1 metodi di genetica molecolare sono usati per diagnosticare la malattia senza dubbio. Questi aiutano a escludere diagnosi differenziali con sintomi simili, ad es. Distrofia Miotonica di tipo 2. La diagnosi può essere supportata da esami elettromiografici (EMG). Nei soggetti colpiti si possono riscontrare schemi tipici di attività spontanea soprattutto sui muscoli distanti dal tronco. È anche importante avere una storia familiare completa, anche al fine di fornire ulteriori consigli alla famiglia.
complicazioni
Con questa malattia, le persone colpite soffrono principalmente di grave debolezza muscolare e sintomi che si verificano negli occhi. Ciò porta alla cataratta e all'opacità del cristallino, in modo che la vista della persona colpita si deteriora in modo significativo. Nel peggiore dei casi, può anche portare alla completa cecità.
La qualità della vita è notevolmente ridotta. I giovani, in particolare, possono sviluppare disturbi psicologici o depressione se hanno problemi visivi improvvisi o se sono ciechi. Inoltre, possono verificarsi problemi cardiaci, in modo che il paziente possa morire di morte cardiaca improvvisa. Non è raro che chi ne soffre soffra di diabete.
La debolezza muscolare limita notevolmente la vita quotidiana delle persone colpite, tanto che in alcuni casi dipendono anche dall'aiuto di altre persone. Alcune attività o sport non possono più essere svolti senza ulteriori indugi. Lo sviluppo dei bambini è significativamente limitato dalla malattia, quindi possono insorgere complicazioni nell'età adulta. Non è possibile trattare questa malattia in modo causale.
Tuttavia, molti reclami possono essere limitati e alleviati in modo che la vita quotidiana diventi sopportabile per la persona interessata. Di regola, non ci sono complicazioni particolari e l'aspettativa di vita del paziente non è limitata dalla malattia.
Quando dovresti andare dal dottore?
La visita di un medico è necessaria non appena la persona interessata ha difficoltà nell'affrontare la vita quotidiana. Una debolezza nella forza muscolare, una diminuzione delle prestazioni fisiche e una perdita di tessuto sono segni di un disturbo della salute. Se le normali attività sportive possono essere praticate solo in misura limitata o per niente, le osservazioni dovrebbero essere discusse con un medico. Devono essere avviati vari esami in modo che la causa possa essere chiarita e un piano di trattamento redatto.
Un ritardo nella tensione muscolare controllata volontariamente e una diminuzione della vista sono preoccupanti. Se la tua vista è offuscata o la lente è opaca, è consigliabile la visita di un medico. Un'irregolarità nella funzione di presa naturale è un segnale di avvertimento del corpo che richiede un'azione. Un aumento del rischio di incidenti e cadute deve essere discusso con un medico in modo che le contromisure possano essere avviate. Disturbi del ritmo cardiaco, palpitazioni o interruzioni del sonno devono essere esaminati più attentamente da un medico.
Se si verificano deficit di concentrazione o di attenzione, o se si nota una riduzione delle prestazioni mentali a causa della menomazione, è necessaria la visita di un medico. Se gli uomini soffrono di diminuzione del desiderio sessuale o se sviluppano una testa calva, è necessario consultare un medico. Se ci sono anche stati di stress emotivo o mentale, la persona colpita è minacciata di conseguenze. Devono essere prevenuti in tempo utile.
Trattamento e terapia
Un trattamento causale di Distrofia miotonica di tipo 1 non può. La terapia si concentra sul sollievo dai sintomi, ad es. attraverso trattamento chirurgico della cataratta, aggiustamento farmacologico per aritmie cardiache o supporto tecnico respiratorio. Il supporto della terapia fisica può ritardare la progressione della distrofia miotonica di tipo 1.
Puoi trovare i tuoi farmaci qui
➔ Medicinali per la debolezza muscolareOutlook e previsioni
Le prospettive di una distrofia miotonica di tipo 1 diagnosticata sono scarse. Sia l'aspettativa di vita che la qualità della vita ne soffrono. La maggior parte dei pazienti non raggiunge nemmeno i 60 anni. Molti di loro muoiono per insufficienza cardiaca o soccombono alle infezioni. Le misure terapeutiche possono spesso alleviare solo marginalmente i sintomi della malattia. Il difetto genetico stesso non è curabile secondo lo stato attuale della scienza. Molte delle persone colpite mostrano segni di Distrofia Miotonica di tipo 1 prima dei 20 anni, altre si rivolgono a un medico solo in età avanzata. All'interno delle famiglie, c'è un aumento del rischio che la malattia venga ereditata.
La sofferenza aumenta continuamente mentre la Distrofia Miotonica di tipo 1 progredisce inesorabilmente nel corso degli anni. A causa della debolezza dei muscoli, le persone colpite trovano sempre più difficile affrontare la vita di tutti i giorni da sole. Avete bisogno di aiuto. Il sistema muscolo-scheletrico si ferma. Dopo un po ', una professione dotta non può più essere esercitata. Gli approcci terapeutici dei farmaci e della fisioterapia perdono sempre più la loro efficacia nel tempo. Non è raro che il declino fisico della Distrofia Miotonica di tipo 1 sia accompagnato da problemi psicologici.
prevenzione
Dal momento che il Distrofia miotonica di tipo 1 Se si tratta di un difetto genetico ereditario, la prevenzione non è possibile.
Dopo cura
La Distrofia Miotonica di tipo 1 è ereditaria. Secondo lo stato attuale della ricerca, una cura non è possibile. La malattia riduce l'aspettativa di vita di circa 50 anni. È consigliabile un follow-up per aiutare a rallentare la progressione della distrofia. Ulteriori obiettivi dell'assistenza post-vendita sono alleviare i sintomi e mantenere la qualità della vita.
Durante il follow-up, viene controllata la tolleranza del farmaco se è stato somministrato al paziente. Le cure di follow-up riguardano principalmente i disturbi fisici. La mobilità degli arti deve essere mantenuta il più a lungo possibile attraverso esercizi appropriati. Anche la psicoterapia di accompagnamento può essere appropriata o addirittura necessaria.
Una qualità della vita inadeguata a causa della distrofia può influenzare l'anima del paziente. Il rischio di depressione è molto alto. Con la psicoterapia c'è l'opportunità di parlare di sentimenti negativi. In una fase avanzata, potrebbe essere necessaria una sedia a rotelle. Durante la cura post-operatoria, il malato impara a utilizzare il dispositivo quotidianamente.
La distrofia miotonica colpisce anche la funzione cardiaca. Un pacemaker contrasta il processo. Il trattamento di follow-up viene eseguito da un cardiologo. Controlla il processo di guarigione dopo l'operazione. Il controllo verrà interrotto quando la guarigione avrà proceduto come previsto.


.jpg)








.jpg)