Al Deficit di α-N-acetilgalattosaminidasi è una malattia da accumulo lisosomiale che si verifica molto raramente. È diviso in una forma giovanile e una adulta.
Che cos'è il deficit di a-N-acetilgalattosaminidasi?

Il deficit di a-N-acetilgalattosaminidasi o Deficit di alfa-N-acetilgalattosaminidasi è anche chiamato in medicina La malattia di Schindler, La malattia di Schindler o La malattia di Kanzaki designato. Ciò che si intende è una malattia da accumulo lisosomiale estremamente rara, la cui eredità è autosomica recessiva. I medici distinguono tra una forma giovanile, che si verifica nei bambini e negli adolescenti, e una forma adulta, di cui soffrono gli adulti.
La forma giovanile di deficit di alfa-N-acetilgalattosaminidasi è chiamata malattia di Schindler, mentre la forma adulta è chiamata malattia di Kanzaki. Ma ci sono anche suddivisioni nella malattia di Schindler di tipo I per la forma giovanile e nella malattia di Schindler di tipo II per la forma adulta. È anche possibile una miscela di entrambe le forme, nota come malattia di Schindler di tipo III.
Tutte e tre le forme della malattia sono oligosaccaridosi (glicoproteinosi), che includono fucosidosi e malattia da accumulo di acido sialico. Il nome malattia di Schindler o malattia di Schindler per il deficit di alfa-N-acetilgalattosaminidasi risale al genetista tedesco Detlev Schindler.
Fu il primo a descrivere la malattia nel 1988. Un anno dopo, il medico giapponese T. Kanzaki descrisse la forma adulta. Ha scoperto nel 1991 che un deficit di alfa-N-acetilgalattosaminidasi era la causa della malattia.
cause
Il deficit di α-N-acetilgalattosaminidasi è causato da una ridotta attività dell'enzima alfa-N-acetilgalattosaminidasi. Le mutazioni missense o nonsense sul gene NAGA, che codifica per l'alfa-N-acetilgalattosaminidasi, sono responsabili del difetto enzimatico.
Il gene NAGA si trova sul locus del gene del cromosoma 22 q11. L'enzima alfa-N-acetilgalattosaminidasi ha il compito di catalizzare la scissione della N-acetilgalattosamina da vari glicolipidi e glicoproteine. Il difetto genetico provoca una ridotta attività dell'enzima, cosicché le sostanze non metabolizzate si accumulano nelle cellule del corpo.
Nella maggior parte dei casi si tratta di proteoglicani, glicosfingolipidi e glicoproteine legate a N o O. L'accumulo di questi provoca danni alla cellula e agli organi colpiti. Il deficit di alfa-N-acetilgalattosaminidasi è così raro che non ci sono informazioni precise sulla sua frequenza.
Finora, solo dodici pazienti di otto famiglie hanno sviluppato la malattia di Schindler nel mondo. I disturbi neurologici non si verificano in tutti i pazienti. Alcuni medici sospettano l'influenza di fattori aggiuntivi per l'insorgenza di sintomi neurologici. Tuttavia, non ci sono ancora prove a sostegno di questa ipotesi.
Puoi trovare i tuoi farmaci qui
➔ Medicinali per il doloreSintomi, disturbi e segni
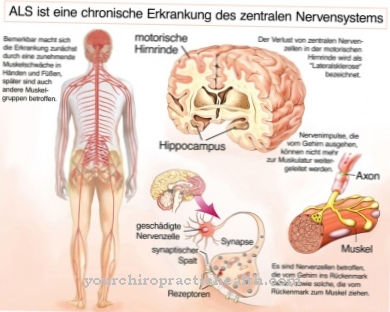
I sintomi del deficit di alfa-N-acetilgalattosaminidasi dipendono dalla particolare forma della malattia. Nel contesto della malattia di Schindler di tipo I, l'ipotonia muscolare progressiva si verifica nel primo anno di vita. Inoltre, i bambini colpiti soffrono di una pronunciata regressione psicomotoria.
Come sintomi sono stati registrati anche disturbi della sequenza di movimento, tetraplegia spastica e crisi cerebrali miocloniche. C'è anche il rischio che il bambino diventi cieco. Se esiste una forma adulta di deficit di alfa-N-acetilgalattosaminidasi, ovvero la malattia di Schindler di tipo 2, si verificano sintomi simili alla malattia di Fabry.
I pazienti hanno angiocheratomi, che sono cambiamenti cutanei benigni. Molti pazienti sono leggermente ritardati mentali. Una forma clinica intermedia è la malattia di Schindler di tipo III Le persone colpite soffrono di disfunzioni neurologiche, convulsioni cerebrali e disabilità mentali.
Sono possibili anche forme meno gravi, associate a lievi disturbi psichiatrici e neurologici, ritardo nello sviluppo del linguaggio o sintomi simili all'autismo.
Diagnosi e corso
La diagnosi di un deficit di alfa-N-acetilgalattosaminidasi è possibile attraverso la rilevazione di una ridotta attività di NAGA. A tale scopo, vengono eseguiti test enzimatici nel plasma sanguigno, leucociti o fibroblasti o linfoblasti coltivati. La rilevazione cromatografica degli oligosaccaridi nelle urine consente anche la diagnosi di carenza enzimatica.
È anche possibile eseguire un'analisi del DNA, ma nella maggior parte dei casi non è necessaria. Durante la gravidanza, la diagnosi prenatale di deficit enzimatico può essere effettuata anche utilizzando un'analisi di mutazione del gene NAGA a seguito di prelievo di villi coriali o amniocentesi. Tuttavia, la rispettiva forma della malattia non può essere determinata.
È importante anche la diagnosi differenziale della sindrome di Fabry, della neurodegenerazione associata alla pantotenato chinasi e della distrofia neuroassonale infantile, che presentano anche difetti enzimatici. Il decorso del deficit di alfa-N-acetilgalattosaminidasi dipende dalla rispettiva forma della malattia.
La malattia di Schindler di tipo I è considerata sfavorevole, mentre la prognosi per gli altri due tipi è più favorevole. Tuttavia, il numero di malati è così piccolo che non si possono fare dichiarazioni significative sull'aspettativa di vita.
complicazioni
I sintomi e le complicazioni del deficit di A-N-acetilgalattosaminidasi dipendono fortemente dalla sua forma.Nella maggior parte dei casi, tuttavia, si verificano disturbi del movimento, quindi le persone colpite sono relativamente limitate nella loro vita quotidiana. Si verificano anche crampi associati a forti dolori.
I bambini in particolare soffrono di gravi disturbi dello sviluppo, che possono portare alla spasticità nel bambino. In alcuni casi, il deficit di A-N-acetilgalattosaminidasi porta alla completa cecità nel paziente. La regressione dell'intelligenza porta al ritardo e quindi alle disabilità.
Questi di solito limitano notevolmente la vita quotidiana del paziente e hanno un effetto negativo sulla qualità della vita. Spesso le persone colpite dipendono dall'aiuto di altre persone. Il ritardo può anche portare a un disturbo della ricerca di parole o un disturbo del linguaggio. Non è possibile trattare il deficit di A-N-acetilgalattosaminidasi causalmente.
Per questo motivo, la persona interessata deve assumere antibiotici. Alcune complicazioni devono anche essere evitate direttamente. La polmonite, ad esempio, è una di queste. Inoltre, il paziente può anche dipendere dalla nutrizione artificiale.
Quando dovresti andare dal dottore?
Il deficit di A-N-acetilgalattosaminidasi deve assolutamente essere trattato da un medico. Questa carenza non scompare da sola e non c'è guarigione spontanea di questa malattia. In ogni caso, un medico deve essere consultato se i genitori notano un calo delle capacità motorie e mentali nel loro bambino o neonato. Reclami con semplici movimenti o processi possono anche indicare la carenza di A-N-acetilgalattosaminidasi e devono essere esaminati da un medico.
Inoltre, varie spasticità sono anche un segno di una malattia. Non è raro che i pazienti soffrano di disabilità intellettive e altri malfunzionamenti. Se questi reclami sono presenti, il trattamento è assolutamente necessario. L'assenza di trattamento può portare a notevoli lamentele e complicazioni in età adulta e quindi ridurre notevolmente la qualità della vita della persona colpita.
Lo sviluppo del linguaggio gravemente ritardato può anche essere un segno di deficit di A-N-acetilgalattosaminidasi e deve quindi essere studiato. Di norma, è possibile consultare un medico generico per determinare il deficit di A-N-acetilgalattosaminidasi. L'ulteriore trattamento dei singoli reclami viene quindi eseguito da uno specialista.
Medici e terapisti nella tua zona
Trattamento e terapia
Una terapia causale per il deficit di alfa-N-acetilgalattosaminidasi non è fattibile. Pertanto, l'approccio medico è limitato al trattamento dei sintomi. Ciò include una dieta ragionevole e l'assunzione di liquidi, la prevenzione delle malattie infettive, che può essere effettuata anche mediante la somministrazione di antibiotici, e il contenimento degli attacchi cerebrali mediante la somministrazione di farmaci antiepilettici.
Ulteriori fasi del trattamento includono misure di fisioterapia per prevenire la polmonite e contratture e per ridurre il dolore o la spasticità con l'aiuto di farmaci. Inoltre, la profilassi dell'aspirazione può avvenire attraverso l'alimentazione artificiale.
Studi recenti hanno dimostrato che il deficit di a-N-acetilgalattosaminidasi è un disturbo del ripiegamento delle proteine, quindi la terapia enzimatica sostitutiva viene discussa come una misura di trattamento ragionevole. La terapia genica è un altro approccio terapeutico concepibile per il trattamento dei disturbi da accumulo lisosomiale. Poiché la malattia di Schindler è ereditata in modo autosomico recessivo, si raccomanda la consulenza genetica delle famiglie colpite.
Outlook e previsioni
Il deficit di A-N-acetilgalattosaminidasi può portare a diversi sintomi. Nella maggior parte dei casi, tuttavia, la carenza porta a un ritardo nello sviluppo psicomotorio. In età adulta, il paziente dipende spesso dall'aiuto di altre persone e non può far fronte alla vita quotidiana da solo. Si verificano anche disturbi del movimento. Questo può essere notato da uno zoppicare o da disturbi della coordinazione.
Nel peggiore dei casi, il paziente può diventare cieco o soffrire di gravi disturbi visivi dovuti al deficit di A-N-acetilgalattosaminidasi. Non è raro che si verifichino disabilità intellettive che riducono notevolmente la qualità della vita della persona interessata. Si verificano anche disturbi del linguaggio e disturbi della ricerca di parole, che possono influenzare la vita. I bambini in particolare sono colpiti da bullismo e prese in giro a causa dei sintomi del deficit di A-N-acetilgalattosaminidasi.
Un trattamento causale del deficit di A-N-acetilgalattosaminidasi non è possibile, quindi il trattamento è principalmente mirato a ridurre i sintomi. Varie misure terapeutiche possono essere utilizzate per ridurre i disturbi dello sviluppo. In molti casi, tuttavia, il paziente dipende da un aiuto esterno nella vita quotidiana. L'aspettativa di vita non è influenzata dalla carenza.
Puoi trovare i tuoi farmaci qui
➔ Medicinali per il doloreprevenzione
Non sono note misure preventive contro il deficit di alfa-N-acetilgalattosaminidasi. La malattia di Schindler è una delle malattie estremamente rare.
Dopo cura
Poiché il deficit di A-N-acetilgalattosaminidasi è una malattia congenita che non può essere trattata in modo causale ma solo sintomatico, il trattamento completo non è possibile. La persona colpita dipende dal trattamento per tutta la vita, motivo per cui le opzioni per la cura successiva sono molto limitate.
Di norma, a causa del deficit di A-N-acetilgalattosaminidasi, il paziente è spesso dipendente dall'assunzione di farmaci e antibiotici. Devono essere prese in considerazione le possibili interazioni con altri farmaci, sebbene gli antibiotici non dovrebbero essere presi insieme all'alcol. Se hai un attacco, dovresti andare in ospedale o chiamare direttamente un medico di emergenza.
Inoltre, i polmoni dovrebbero essere risparmiati per evitare l'infiammazione. Pertanto, i pazienti con deficit di A-N-acetilgalattosaminidasi non devono fumare in nessuna circostanza. Una dieta sana e uno stile di vita generalmente sano hanno un effetto molto positivo sul decorso della malattia.
Se il paziente desidera avere figli, la consulenza genetica è utile per prevenire la trasmissione della malattia ai bambini. Poiché i pazienti spesso soffrono di mobilità ridotta, la fisioterapia è utile per alleviarla. Molti esercizi possono essere eseguiti anche a casa tua.
Puoi farlo da solo
Un deficit esistente di A-N-acetilgalattosaminidasi è una malattia incurabile. Il trattamento medico è quindi essenziale. Le misure di auto-trattamento possono essere basate solo sui sintomi per rendere più facile la vita di tutti i giorni. È importante che i genitori osservino il bambino da vicino.
I genitori possono aiutare al meglio i loro figli attraverso la comprensione e la pazienza, l'amore e la cura. Inoltre, i trattamenti di terapia occupazionale e logopedia si basano sempre su un duplice concetto: terapia in loco con lo specialista ed esecuzione degli esercizi a casa. I genitori dovrebbero essere attivi qui in modo coerente e con pazienza. Deve essere assicurata una dieta ricca di vitamine e minerali, un regolare esercizio fisico e molta aria fresca. Questo rafforza il sistema immunitario e riduce il rischio di infezione. Se la persona ha la tendenza ad avere crisi epilettiche, non dovrebbe essere sola per molto tempo e dovrebbe essere controllata per vedere se può ferirsi se si verifica una crisi.
Il più delle volte, i pazienti non sono in grado di affrontare da soli la vita di tutti i giorni e richiedono cure costanti. Se i genitori non possono permetterselo, specialmente con i bambini più grandi e gli adulti, non dovrebbero aver paura di cercare aiuto. Questo può assumere la forma di un'infermiera o di una sistemazione in una struttura adeguata. Se vuoi ancora avere figli, si consiglia una consultazione con uno specialista, poiché la malattia è ereditaria.

.jpg)
.jpg)

.jpg)

.jpg)
.jpg)