Il Sindrome di Jirásek-Zuelzer-Wilson, anche Sindrome di Zuelzer-Wilson chiamato aganglionosi dell'intestino. Anche nell'infanzia, i pazienti soffrono di problemi di defecazione e gonfiore.
Che cos'è la sindrome di Jirásek-Zuelzer-Wilson?

© Sebastian Kaulitzki - stock.adobe.com
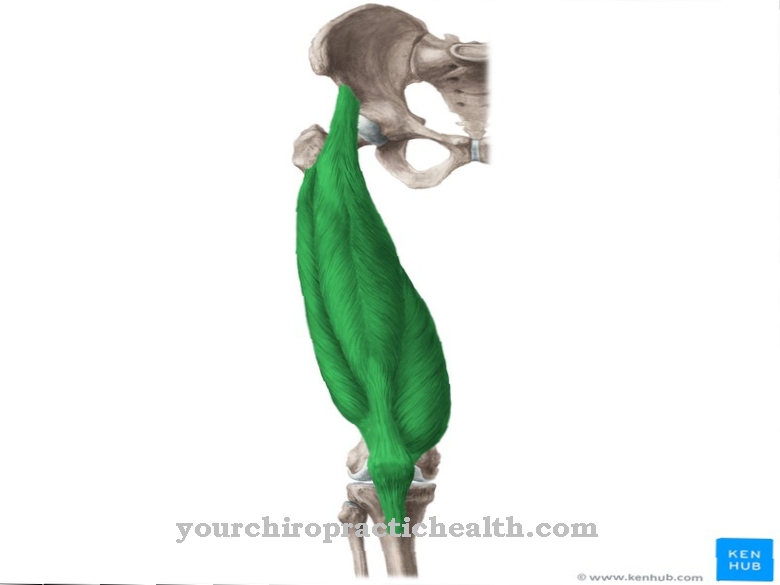
Il Sindrome di Jirásek-Zuelzer-Wilson prende il nome dai dottori Wolf William Zülzer, James Leroy Wilson e Arnold Jirásek. Per prima cosa hanno descritto la forma congenita e rara di aganglionosi. L'aganglionosi è la mancanza congenita di neuroni nella parete dell'intestino. I neuroni sono generalmente assenti nel retto e / o nell'intestino crasso.
Le cellule gangliari del plesso di Auerbach (plesso mioentico) o del plesso di Meissner (plesso sottomucoso) sono colpite dalla malattia. Entrambi i plessi nervosi fanno parte del sistema nervoso intramurale o enterico e quindi appartengono al sistema nervoso vegetativo. I gangli del plesso di Meissner si trovano nella sottomucosa, nello strato tra lo strato muscolare e la mucosa dell'intestino.
Il plesso di Meissner controlla la secrezione delle ghiandole gastriche e intestinali indipendentemente dal sistema nervoso centrale. I movimenti epiteliali dell'intestino e i processi immunologici sono controllati anche dal plesso sottomucoso. Il plesso sottomucoso è strettamente connesso al plesso mioenterico. Il plesso mioenterico si trova tra i muscoli circolari e longitudinali nella parete dell'apparato digerente.
Controlla la peristalsi e la motilità dello stomaco, dell'intestino e dell'esofago. Proprio come il plesso sottomucoso, agisce indipendentemente dal sistema nervoso centrale. Tuttavia, può essere influenzato nella sua attività tramite il sistema nervoso simpatico e parasimpatico. A differenza della malattia di Hirschsprung, la sindrome di Jirásek-Zuelzer-Wilson colpisce le strutture nervose in tutto il colon.
cause
L'aganglionosi provoca un'eccessiva formazione di cellule nelle cellule nervose a monte. Di conseguenza, anche il neurotrasmettitore acetilcolina viene sempre più sintetizzato e rilasciato. Ciò si traduce in una stimolazione costante dei muscoli circolari dell'intestino, in modo che vi sia una contrazione permanente della rispettiva sezione intestinale. La sovraeccitazione dei muscoli circolari restringe il tubo intestinale, provocando un'ostruzione intestinale.
L'intestino non può più essere svuotato adeguatamente. Si sviluppa grave stitichezza con accumulo di feci. Le feci si espandono davanti al segmento ristretto e può svilupparsi un megacolon.
La malattia è probabilmente basata su un difetto nell'immigrazione dei neuroblasti. I neuroblasti sono cellule progenitrici di cellule nervose che possono dividersi. Inoltre, i neuroblasti immigrati hanno disturbi della maturazione. Anche una temporanea riduzione del flusso sanguigno nell'intestino o infezioni virali nell'utero sono possibili cause della sindrome di Jirásek-Zuelzer-Wilson.
Poiché la malattia si manifesta nelle famiglie, si presume una predisposizione genetica. Nella malattia di Hirschsprung, un'altra aganglionosi, sono state trovate mutazioni nel gene dell'endotelina-3 (EDN3) e nel gene del recettore dell'endotelina (EDNRB). La malattia di Hirschsprung e la sindrome di Jirásek-Zuelzer-Wilson si verificano più frequentemente quando i parenti si sposano. Pertanto, la malattia è abbastanza comune, specialmente tra gli Amish negli Stati Uniti.
Sintomi, disturbi e segni
I primi sintomi della sindrome di Jirásek-Zuelzer-Wilson compaiono solitamente nei primi giorni dopo la nascita. Una mancanza di rimozione del meconio è caratteristica. Il meconio è anche popolarmente noto come passo bambino. È la prima sedia del neonato. Questo contiene epitelio esfoliato, bile ispessita, capelli e cellule della pelle e viene escreto nelle prime 24-48 ore dopo la nascita.
Un'ostruzione intestinale da meconio, che è in realtà tipica della malattia della fibrosi cistica, può indicare la sindrome di Jirásek-Zuelzer-Wilson. La sindrome di Jirásek-Zuelzer-Wilson si verifica piuttosto raramente negli adulti. Le persone colpite soffrono di stitichezza cronica. Il più delle volte, l'aganglionosi colpisce solo una parte molto breve dell'intestino nei pazienti adulti. I sintomi non sono così pronunciati che la diagnosi viene fatta molto tardi.
Diagnosi e decorso della malattia
La diagnosi sospetta di solito può essere formulata sulla base del quadro clinico. Per confermare la diagnosi, viene eseguita, tra le altre cose, una manometria. Viene misurata la pressione nell'area tra l'ano e il retto. Una biopsia di aspirazione dal rivestimento del retto può essere eseguita anche in anestesia generale.
La mancanza di cellule gangliari può quindi essere rilevata dall'esame patologico delle cellule bioptiche. Una diagnosi radiografica con un clistere intestinale di agente di contrasto non è significativa. Questo esame può solo valutare l'entità dei cambiamenti. La procedura è importante anche nella preparazione per la biopsia seriale.
Sulla base dell'esame chimico degli enzimi si può rilevare un'aumentata attività dell'acetilcolinesterasi nei primi mesi di vita e quindi una malnervazione colinergica della mucosa intestinale. Un'altra indicazione diagnostica della sindrome di Jirásek-Zuelzer-Wilson è la mancanza della proteina calretinina. Questo è normalmente espresso nelle cellule gangliari.
complicazioni
La qualità della vita è notevolmente limitata e ridotta dalla sindrome di Jirásek-Zuelzer-Wilson. Nella maggior parte dei casi, i sintomi della sindrome di Jirásek-Zuelzer-Wilson compaiono immediatamente dopo la nascita. I bambini soffrono di problemi digestivi. Con il progredire della malattia si verifica anche stipsi cronica.
Questa stitichezza può ancora essere presente in età adulta e quindi limitare in modo significativo la vita quotidiana della persona interessata. Non è raro che disturbi di stomaco permanenti portino a disturbi psicologici o depressione grave. Lo stress può anche aumentare e intensificare questi sintomi. La sindrome di Jirásek-Zuelzer-Wilson può essere trattata relativamente bene rimuovendo la parte dell'intestino interessata.
Non ci sono particolari complicazioni o reclami. Allo stesso modo, l'infiammazione dello stomaco e dell'intestino deve continuare a essere combattuta. In alcuni casi è quindi necessaria un'uscita artificiale nell'intestino, ma questa non rimane permanente. Dopo il trattamento di solito non ci sono più disturbi o sintomi. La sindrome di Jirásek-Zuelzer-Wilson non riduce l'aspettativa di vita delle persone colpite. Non è raro che i trattamenti vengano ripetuti.
Quando dovresti andare dal dottore?
Poiché la sindrome di Jirásek-Zuelzer-Wilson viene diagnosticata più spesso nei neonati o nei bambini, i genitori e le ostetriche in particolare dovrebbero prestare particolare attenzione alle dimissioni del bambino. Tuttavia, se gli adulti notano cambiamenti improvvisi nei movimenti intestinali, anche loro dovrebbero sottoporsi a una visita medica completa. Se i bambini hanno problemi a defecare, consultare un medico. Se non c'è movimento intestinale per diversi giorni, questo è un avvertimento. Per evitare ulteriori malattie batteriche o infiammazioni, consultare un medico il prima possibile.
I neonati sono spesso in ospedale per i primi giorni di vita e sono sotto costante controllo medico. Le prime irregolarità vengono solitamente scoperte dal personale infermieristico del reparto neonatale, in modo che i genitori del bambino non debbano intraprendere alcuna azione. Se le prime feci del neonato contengono epidermide o capelli, sono necessari ulteriori esami.
Se i sintomi compaiono poche settimane o mesi dopo il parto, consultare un medico non appena si sviluppa la stitichezza. Spesso si sviluppa uno stomaco gonfio, che indica discrepanze esistenti. Se il bambino soffre o ha problemi comportamentali, è necessaria la visita di un medico. Un medico è necessario in caso di rifiuto di mangiare, comportamento apatico o aggressivo, nonché comportamento piagnucoloso o urlante.
Trattamento e terapia
Se la sindrome di Jirásek-Zuelzer-Wilson si verifica nel neonato, di solito deve essere posizionato almeno temporaneamente un ano artificiale. In alternativa, l'intestino può essere risciacquato o svuotato il più completamente possibile con un tubo intestinale. Tuttavia, la sezione dell'intestino interessata di solito deve essere rimossa chirurgicamente.
Se l'aganglionosi colpisce solo un segmento molto corto dell'intestino, il muscolo contratto può essere inciso. Questa procedura è anche nota come miectomia dello sfintere. A seconda dell'entità della malattia e dell'esperienza della clinica curante, per il trattamento della sindrome di Jirásek-Zuelzer-Wilson vengono utilizzate procedure chirurgiche laparoscopiche, transanale o aperte.
Se la condizione non viene trattata abbastanza presto, può svilupparsi l'enterocolite. L'enterocolite è un'infiammazione acuta del tratto gastrointestinale. La causa della malattia è una combinazione di parete intestinale danneggiata e infezione. Il deterioramento dei tessuti si verifica come risultato di questi due fattori.
In caso di gravi danni, la parete intestinale può perforarsi, in modo che il contenuto intestinale entri nella cavità addominale e causi l'infiammazione del peritoneo (peritonite). Ne può derivare una sepsi pericolosa per la vita.
Puoi trovare i tuoi farmaci qui
➔ Medicinali per costipazione e problemi intestinaliOutlook e previsioni
Le prospettive di guarigione o di sollievo chirurgico sono fosche in presenza della sindrome di Jirásek-Zuelzer-Wilson. È vero che le terapie e le operazioni possono portare un certo sollievo. Tuttavia, rimane difficile impostare chirurgicamente il passaggio intestinale in modo che funzioni senza intoppi. Le numerose conseguenze della sindrome di Jirásek-Zuelzer-Wilson non possono essere del tutto eliminate.
Lo sviluppo di un megacolon o un'ostruzione intestinale sono due delle possibili conseguenze della sindrome di Jirásek-Zuelzer-Wilson. Questo è probabilmente causato geneticamente. Pertanto si verifica già nell'infanzia. La parte interessata dell'intestino di solito deve essere rimossa. Tuttavia, rimangono abbastanza sintomi in modo che depressione, menomazioni della vita e disturbi mentali non siano di rado il risultato della sindrome di Jirásek-Zuelzer-Wilson.
La durata della vita non è ridotta nella sindrome di Jirásek-Zuelzer-Wilson. Ma la stitichezza costante, l'infiammazione gastrica o un ano artificiale temporaneo mettono a dura prova la qualità della vita. L'enterocolite è probabile con il trattamento medico tardivo. Ciò grava anche sulle persone colpite. Nel peggiore dei casi, segue la peritonite, che se non trattata può trasformarsi in sepsi pericolosa per la vita.
La rarità di questa malattia è problematica per le persone colpite. Pertanto, non ci sono quasi nessun gruppo di auto-aiuto e nessuno scambio, a meno che non ci siano pazienti affetti anche all'interno della famiglia. Di conseguenza, le persone colpite si sentono spesso isolate. Molti soffrono di stress emotivo e stress. Dopo che l'intero colon è stato rimosso chirurgicamente, lo stress è ancora maggiore.
prevenzione
La sindrome di Jirásek-Zuelzer-Wilson non può essere prevenuta perché i meccanismi esatti con cui si sviluppa sono sconosciuti.
Dopo cura
Nella maggior parte dei casi di sindrome di Jirásek-Zuelzer-Wilson, la persona colpita ha pochissime o addirittura nessuna misura speciale e opzioni per le cure di follow-up disponibili, quindi con questa malattia deve essere fatta una diagnosi rapida e soprattutto precoce della malattia per prevenire ulteriori complicazioni o disagio.
L'auto-guarigione non può verificarsi con la sindrome di Jirásek-Zuelzer-Wilson, quindi una diagnosi precoce ha sempre un effetto positivo sull'ulteriore decorso della malattia. Nella maggior parte dei casi, questa malattia richiede un intervento chirurgico. Dopo tale operazione, la persona colpita dovrebbe assolutamente riposare e prendersi cura del proprio corpo, anche se dovrebbero essere evitate attività fisiche o stressanti.
Allo stesso modo, i cibi grassi dovrebbero essere evitati. Poiché la sindrome può danneggiare anche altri organi interni, il paziente deve sottoporsi a regolari esami del corpo per verificare le condizioni degli organi interni. La sindrome può anche ridurre l'aspettativa di vita delle persone colpite. Spesso è necessario anche l'aiuto e il sostegno della propria famiglia o dei propri amici, in particolare per prevenire disturbi psicologici o depressione.
Puoi farlo da solo
I pazienti con ano artificiale e colectomia (asportazione chirurgica dell'intero intestino crasso) potrebbero dover imparare di nuovo a trattenere la voglia di defecare o allungare gli intervalli. Medici e cliniche forniscono informazioni su come può svolgersi tale formazione, ma in realtà questo è il dominio dei fisioterapisti. Conoscono il corpo, i suoi muscoli e le fasce e sanno quali esercizi rafforzano quali muscoli e gruppi di muscoli.
Le escrezioni altamente fluide dall'ano come muco o sangue, ma anche feci molto aggressive, stressano la pelle; dopo un po 'diventa dolorante, screpolato e non guarisce più. Il cibo addensato con psillio o bucce di psillio può impedirlo. I cosiddetti raccoglitori fecali sono incollati tra i glutei e raccolgono le escrezioni. I raccoglitori di feci sono prescritti di default per i pazienti sdraiati, ma poiché sono delicati sulla pelle, sono un auto-aiuto che vale la pena considerare. Fare attenzione quando si allenta la superficie adesiva: qui possono verificarsi difetti della pelle.
La sindrome di Jirásek-Zuelzer-Wilson è una malattia estremamente rara; I gruppi di auto-aiuto per questa malattia sono rari e quindi difficili da trovare. La compagnia di assicurazione sanitaria può essere un primo punto di contatto qui. Anche se non si conosce alcun gruppo, i dipendenti delle compagnie di assicurazione sanitaria di solito conoscono approcci alternativi a come trovare un tale gruppo.

.jpg)

.jpg)

.jpg)

.jpg)

.jpg)
.jpg)