Il Sindrome di Holt-Oram è una sindrome malformativa che è principalmente associata a difetti cardiaci e anomalie dei pollici ed è causata da una mutazione. Nella maggior parte dei casi, la mutazione causale si verifica sporadicamente e quindi corrisponde a una nuova mutazione. La correzione chirurgica del difetto cardiaco è il fulcro della terapia.
Sindrome di Holt-Oram?

© SmirkDingo - stock.adobe.com
Le sindromi malformative congenite con coinvolgimento predominante degli arti sono un gruppo di malattie che comprende varie deformità delle braccia e delle gambe. Uno di loro è questo Sindrome di Holt-Oram, chiamato anche Sindrome cuore-mano è conosciuto. La sindrome è associata al gruppo delle displasie atriodigitali, che comprendono malattie congenite con malformazioni degli arti superiori e del cuore.
Oltre alla sindrome di Holt-Oram, a questo gruppo di malattie appartengono la sindrome cuore-mano di tipo 2, la sindrome cuore-mano di tipo 3 e la sindrome cuore-mano di tipo sloveno. La sindrome di Holt-Oram è stata descritta per la prima volta nel 1960. Il pediatra britannico Holt e il cardiologo Samuel Oram sono i primi a descrivere la malattia. La sindrome di Holt-Oram Questa è una malattia relativamente rara ed è associata a una prevalenza media di una persona affetta su 100.000 persone.
La causa delle malformazioni delle mani e del cuore nella sindrome di Holt-Oram risiede nella genetica. I sintomi della sindrome sono tanto vari quanto le sue cause sospette. Le malformazioni nella malattia si concentrano sul cuore e sul pollice.
cause
Sebbene la sindrome di Holt-Oram sia una malattia genetica e congenita, non c'è quasi alcuna frequenza familiare nei casi finora documentati. Sebbene la sindrome sembri essere ereditata con modalità di trasmissione autosomica dominante nei singoli casi, gran parte della documentazione del caso suggerisce la sua presenza sporadica. Circa l'85% dei casi documentati sembra essere dovuto a una nuova mutazione.
La causa principale della sindrome di Holt-Oram risiede nelle mutazioni genetiche nel locus genico 12q23-24.1. Il cosiddetto gene TBX5, che si trova sul cromosoma 12 ed è una proteina coinvolta nell'arto e nell'arto, si trova in questo locus genico Sviluppo del cuore codificato. Le funzioni esatte della proteina non sono state ancora chiarite. Inoltre non è stato ancora stabilito se fattori esterni come l'esposizione a tossine o la malnutrizione nella madre durante la gravidanza favoriscano la mutazione del gene TBX5.
Le mutazioni nel gene possono essere rilevate in almeno fino a 70 persone su 100 pazienti con sindrome di Holt-Oram. Tuttavia, la scienza presume che anche le anomalie in altri geni possano causare i sintomi della sindrome. Ad esempio, la sindrome da malformazione è associata alla polisindattilia del pollice tripartita.
Sintomi, disturbi e segni
I pazienti con sindrome di Holt-Osram soffrono di un complesso di malformazioni che colpiscono principalmente il pollice e il cuore. Sebbene la localizzazione delle malformazioni del paziente sia comune, sono possibili diversi tipi di malformazioni sul cuore e sul pollice. Il quadro clinico è quindi estremamente vario.
I difetti cardiaci possono manifestarsi, ad esempio, sotto forma di un difetto del setto ventricolare, un difetto del setto atriale, un'aritmia cardiaca o un disturbo della conduzione. Le anomalie scheletriche possono corrispondere a malformazioni di riduzione dei pollici, ma si verificano anche anomalie come la mancata applicazione del raggio.
Molti pazienti con la sindrome soffrono anche di sinostosi radioulnari e anomalie delle costole, della scapola o della clavicola. Inoltre, la sindrome di Holt-Oram è associata a pectus carinatum e scoliosi. La maggior parte delle persone colpite soffre anche di sindattilia delle falangi delle dita delle mani o dei piedi. In singoli casi questi sintomi sono associati all'ipertelorismo.
Diagnosi e decorso della malattia
La sindrome di Holt-Oram è spesso mal diagnosticata. In una diagnosi differenziale, il medico deve differenziare il complesso dei sintomi dalla sindrome di Okihiro dopo mutazioni geniche nel gene SALL4 sul cromosoma 20, che è associato alle stesse malformazioni del braccio e difetti cardiaci. Ciò che è particolarmente rilevante nella diagnosi differenziale è che i pazienti con sindrome di Okihiro di solito hanno un'anomalia di Duane.
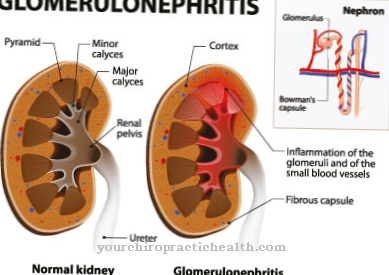
Strizzano gli occhi, sono spesso colpiti da malformazioni renali e hanno disturbi dell'udito, anomalie del piede o malformazioni dell'orecchio. La sindrome Trombocitopenia-Assenza-Radio deve essere differenziata anche dalla sindrome di Holt-Oram, che si ottiene principalmente attraverso la diagnostica di laboratorio. Altri quadri clinici con un quadro clinicamente simile sono l'anemia di Fanconi e la sindrome di Pallister Hall.
L'aspettativa di vita dei pazienti con sindrome di Holt-Oram non è inferiore alla media. Solo nei casi più gravi esiste un difetto cardiaco difficilmente curabile che rende la prognosi sfavorevole.
complicazioni
La sindrome di Holt-Oram causa una serie di diverse malformazioni e deformità nel paziente, che possono rendere difficile la vita e la vita di tutti i giorni. Soprattutto, il cuore è colpito dalle malformazioni, quindi il paziente soffre di un difetto cardiaco. C'è anche un'aritmia cardiaca dalla quale la persona colpita può morire nel peggiore dei casi.
Anomalie si verificano anche sui pollici, in modo che anche determinati movimenti o processi nella vita di tutti i giorni siano resi più difficili. Non è raro che le deformità del corpo portino a prese in giro e bullismo di altri bambini, il che può portare a disturbi psicologici e depressione in molti pazienti. Non è raro che si verifichino disturbi renali, che nel peggiore dei casi possono portare a insufficienza renale.
Inoltre, le persone colpite soffrono anche di disabilità visive e uditive. Di regola, l'aspettativa di vita rimane invariata a causa della sindrome di Holt-Oram, a condizione che non vi sia alcun difetto cardiaco che porti alla morte. Un trattamento causale della sindrome di Holt-Oram di solito non è possibile, quindi solo i sintomi possono essere trattati. In molti casi è necessario anche un supporto psicologico.
Quando dovresti andare dal dottore?
La sindrome di Holt-Oram viene solitamente diagnosticata subito dopo la nascita. A seconda della gravità delle malformazioni, il bambino affetto potrebbe aver bisogno di ulteriori esami medici. In linea di principio, i difetti cardiaci devono essere trattati tempestivamente per ridurre il rischio di gravi malattie secondarie. Se si sviluppano complicazioni come aritmie cardiache o segni di difetto del setto interatriale, è necessario consultare un medico. Un medico dovrebbe anche essere consultato con anomalie del pollice scheletrico.
I genitori dei bambini affetti devono consultare attentamente il proprio medico e informarli di eventuali sintomi insoliti. Poiché la sindrome di Holt-Oram è una malattia ereditaria, non è possibile alcun trattamento causale. I pazienti possono quindi dover essere trattati per tutta la vita, a seconda di quali malformazioni si verificano e quale sia la costituzione del paziente. Poiché questo spesso causa anche disturbi emotivi, è indicato il supporto psicologico di accompagnamento. I bambini che soffrono di bullismo o presa in giro dovrebbero cercare consulenza terapeutica con i loro genitori.
Medici e terapisti nella tua zona
Terapia e trattamento
Non sono disponibili trattamenti causali per i pazienti con sindrome di Holt-Oram. C'è speranza per una trattabilità causale in futuro, poiché la terapia genica è attualmente oggetto di ricerca medica. Tuttavia, fino a quando questo tipo di terapia non raggiunge la fase clinica, la sindrome di Holt-Oram rimane una malattia incurabile.
Al momento, sono disponibili solo opzioni di terapia sintomatica per il trattamento dei pazienti. La terapia si basa sui sintomi nel singolo caso. La correzione precoce del difetto cardiaco è di particolare rilevanza. Questa correzione viene eseguita chirurgicamente. Nel caso di un difetto del setto di Vohof, la procedura chirurgica mira, ad esempio, a chiudere il difetto in questione. Lo stesso vale per un difetto del setto ventricolare.
Le correzioni alle malformazioni alle estremità sono inizialmente di secondaria importanza. Dopo aver corretto con successo il difetto cardiaco, le procedure chirurgiche ricostruttive possono ripristinare i raggi mancanti e le sindattilia separate. Una scoliosi esistente viene solitamente combattuta sotto cure fisioterapiche. In casi particolarmente gravi, può essere necessario un intervento chirurgico per impiantare una protesi costale in titanio.
Nella maggior parte dei casi, non è richiesto alcun intervento per il pectus carinatum. Per ragioni psicologiche, invece, il torace può essere rimodellato chirurgicamente, ad esempio seguendo la procedura di Nuss.
Outlook e previsioni
La prognosi per la sindrome di Holt-Oram è favorevole. Sebbene ci sia un difetto genetico, può essere adeguatamente trattato con le attuali opzioni mediche. L'aspettativa di vita di qualcuno con la sindrome nella maggior parte dei casi non è inferiore alla media. In caso di malformazione grave, possono esserci perdite significative nell'aspettativa di vita. La prognosi è chiaramente peggiorata in questi pazienti. L'attività del cuore è limitata e può portare a una fine prematura della vita.
Tuttavia, la maggior parte dei pazienti può essere trattata bene e con successo. Sebbene non esista una cura a causa del difetto genetico presente, ci sono buone prospettive per varie opzioni di correzione. L'attività del cuore viene regolata e, se possibile, completamente corretta in un intervento chirurgico. Sebbene possa esserci una compromissione permanente nel modo di vivere rispetto alle persone sane, grazie al trattamento si ottiene una buona qualità della vita.
Anomalie fisiche o malformazioni vengono modificate in una fase successiva. Di solito, dopo che la fase di crescita del bambino è completa, viene avviata una correzione necessaria o desiderata delle malformazioni esistenti. Se i disturbi nel processo di sviluppo portano a menomazioni significative, durante l'infanzia vengono prese misure correttive per alleviare i sintomi. A causa dei cambiamenti ottici, il paziente può subire conseguenze psicologiche. Ciò peggiora la prognosi generale.
prevenzione
Finora la sindrome di Holt-Oram non può essere prevenuta perché i fattori di influenza esterni non sono stati chiariti in modo definitivo.
Dopo cura
Poiché la sindrome di Holt-Oram è una malattia congenita, non può essere completamente curata. Pertanto, le misure o le possibilità di assistenza di follow-up sono molto limitate, per cui la persona colpita dipende principalmente dalla diagnosi precoce e dal successivo trattamento. Se il paziente oi genitori desiderano avere figli, viene fornita consulenza genetica per evitare che questa sindrome si ripresenti.
Il trattamento della sindrome di Holt-Oram è principalmente finalizzato al trattamento del difetto cardiaco. Questo viene corretto da una procedura chirurgica, in base alla quale il paziente deve riprendersi e riposare dopo la procedura. Lo sforzo o l'attività fisica dovrebbero essere evitati. Non è raro che sia richiesto un trattamento fisioterapico e molti degli esercizi possono essere eseguiti anche a casa propria.
Le persone colpite a volte dipendono dall'aiuto e dal sostegno della propria famiglia e dei propri amici. Questo può anche prevenire disturbi psicologici o depressione. Inoltre, uno stile di vita sano con una dieta sana ha un effetto molto positivo sul decorso della sindrome di Holt-Oram.
Puoi farlo da solo
La sindrome di Holt-Oram non può essere prevenuta e inoltre non può essere trattata con mezzi di autoaiuto. Con questa sindrome, le persone colpite dipendono sempre da un'operazione per trattare il difetto cardiaco al fine di estendere l'aspettativa di vita del paziente. Prima viene riconosciuta la sindrome, maggiori sono le possibilità di un decorso positivo della malattia. Anche le altre malformazioni del corpo devono essere corrette chirurgicamente.
Poiché molte delle persone colpite soffrono anche di disturbi psicologici o di complessi di inferiorità associati a questa sindrome, dipendono dal trattamento psicologico. Tuttavia, le conversazioni con altri pazienti, i tuoi genitori o amici possono anche rafforzare la fiducia in se stessi del paziente e quindi alleviare i disturbi psicologici. Inoltre, alcuni pazienti dipendono dall'aiuto dei loro simili nella vita di tutti i giorni, per cui l'assistenza calda ha un effetto molto positivo sul decorso della sindrome di Holt-Oram.
Poiché la malattia può colpire anche gli organi interni, i pazienti dipendono da esami e controlli regolari da parte di vari medici. Questo può prevenire problemi ai reni, per esempio. I bambini affetti dovrebbero essere informati sulle conseguenze e le complicazioni della malattia.




.jpg)
.jpg)


.jpg)